
- Plasmids Were Discovered in 1952 as Agents of "Genetic Exchange" Between Bacteria
- In the Last 50 Years, There Have Been Over 200,000 Plasmid Publications, Averaging 15 Per Day During 1990-2020
- Plasmids Provide the Starting Point for IVT mRNA Production
TriLink recently introduced plasmid manufacturing services as advantageous entry points for the in vitro transcription (IVT) template production of your mRNA, and this prompted the Zone to provide a series of blogs exploring plasmid biology and manufacturing. This initial (Part 1) blog offers some introductory comments on the discovery of plasmids that paved the way for recombinant DNA and early IVT mRNA technologies. Part 2, which will be posted in July of 2021, transitions to discussing the key elements required to perfect plasmid production in the context of IVT mRNA manufacturing by TriLink.
To learn more, register for the upcoming June 15, 2021, TriLink mRNA Basics webinar: Getting Started with GMP mRNA Manufacturing.
The Nature and Discovery of Plasmids
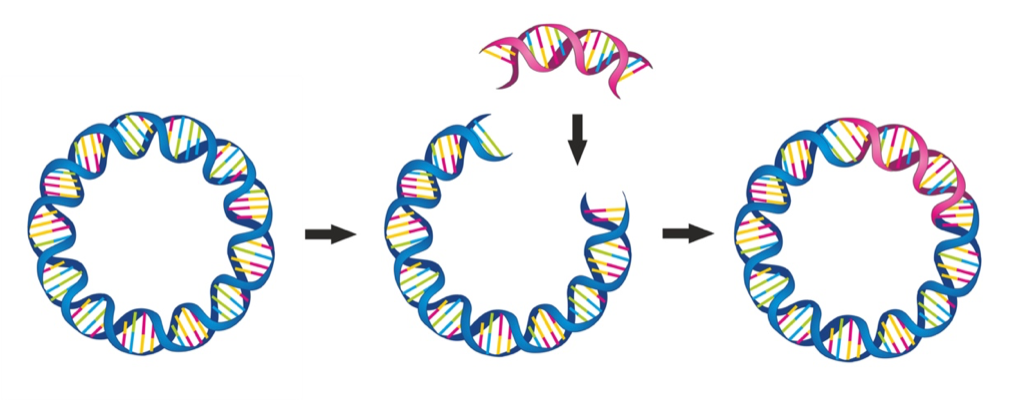
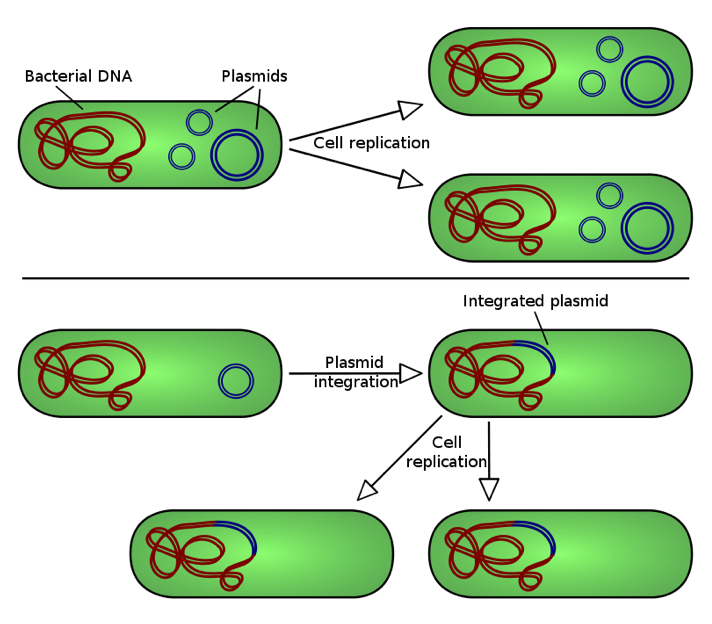
Textbook definitions of a plasmid usually read something like this: a plasmid is a relatively small, extrachromosomal DNA molecule within a cell that is physically separated from chromosomal DNA and can replicate independently. Although most commonly found as small, circular, double-stranded DNA (dsDNA) molecules in bacteria, plasmids are sometimes also present in archaea and eukaryotic organisms. As depicted here, there are two ways plasmids integrate into a host bacterium: non-integrating plasmids replicate as shown in the top figure, whereas episomes, shown in the lower figure, can integrate into the host chromosome. In this blog, we will only consider non-integrating plasmids.
In nature, plasmids often carry genes that benefit the survival of the organism, conferring selective advantages like antibiotic resistance. While chromosomes are comparatively large and contain all the essential genetic information required for life under normal conditions, plasmids only contain additional genes that may be useful in certain situations or conditions.
Introduced in 1952 by Joshua Lederberg, the term plasmid refers to "any extrachromosomal hereditary determinant," and it relates to a definitive study of "genetic exchange" in Salmonella typhimurium reported earlier that year by Lederberg and his graduate student Norton Zinder. For this work, Lederberg was awarded a share of the 1958 Nobel Prize in Physiology or Medicine at only 33 years of age.
According to Wikipedia, the term initially encompassed any bacterial genetic material that exists extrachromosomally for at least part of its replication cycle. Because that description includes bacterial viruses, the definition of plasmid was refined over time, becoming limited to genetic elements that reproduce autonomously. Later in 1968, the term plasmid was adopted to define extrachromosomal genetic elements and distinguish from viruses. The definition was narrowed to genetic elements that can replicate autonomously and exist exclusively or predominantly outside the chromosome.
While concluding this brief introduction to plasmids, it is worth noting that, following the above-mentioned 1952 publications by Lederberg, nearly 200,000 publications have been indexed to the term plasmid(s) in the PubMed database of scientific, technical, biomedical, and health journals, books, etc. This huge number does not include the many number patents related to plasmids, especially in biotechnology. As shown by the chart here, the number of publications rose sharply between 1970 and 1980.
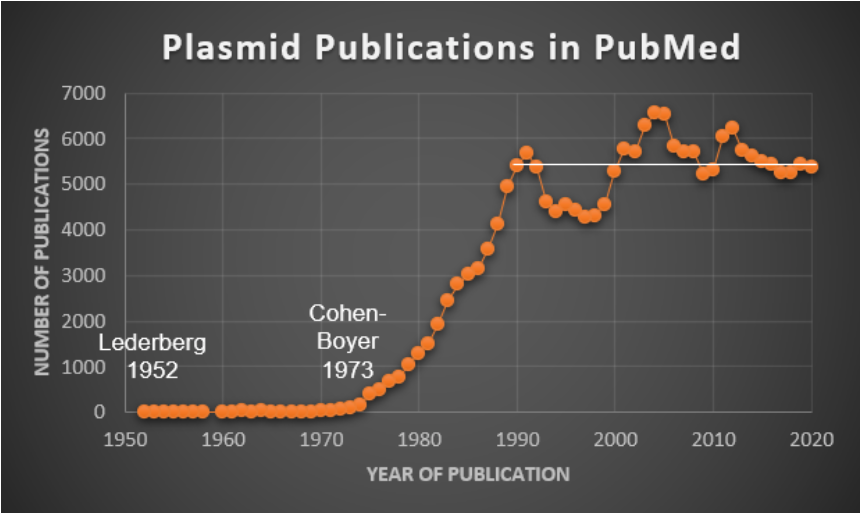
As discussed in the next section, this upward trend is associated with the emergence of plasmid-based recombinant DNA technology for basic and applied purposes. Additionally, for each year from 1990 to 2020, approximately5,000 plasmid-related publications have been released, as indicated by the white line. This translates to an average of 15 publications every day, each week, nonstop.
The Birth of Recombinant DNA Technology
In 1973, Stanley Cohen at Stanford University, in collaboration with others, published a paper in the prestigious Proc. Natl. Acad. Sci. journal titled Construction of Biologically Functional Bacterial Plasmids In Vitro. With more than 2,300 citations in Google Scholar, this landmark paper, coauthored by Herbert Boyer at the University of California, San Francisco, is associated with the genesis of recombinant DNA technology, the birth of genetic engineering, and the beginning of commercial biotechnology (see Cohen-Boyer patent). Cohen would later share the 1968 Nobel prize for Physiology or Medicine for his discovery of epidermal growth factor.

In brief, the now famous 1973 Cohen, Boyer, et al. publication described "[t>
he construction of new plasmid DNA species by in vitro joining of restriction endonuclease-generated fragments of separate plasmids." Newly constructed plasmids that are inserted into Escherichia coli (E. coli) by transformation were shown to be biologically functional replicons that possessed genetic properties and nucleotide base sequences from both parent DNA molecules.
The prescient concluding paragraph understatedly reads:
"The general procedure described here is potentially useful for insertion of specific sequences from prokaryotic or eukaryotic chromosomes or extrachromosomal DNA into independently replicating bacterial plasmids. The antibiotic resistance plasmid pSC101 constitutes a replicon of considerable potential usefulness for the selection of such constructed molecules, since its replication machinery and its tetracycline resistance gene are left intact after cleavage by the EcoRI endonuclease."
Indeed, these selection features are still widely used today.
Creation of the Recombinant DNA Advisory Committee (RAC)
Concurrent with the seminal Cohen-Boyer publication and related developments, serious safety concerns were voiced in a published letter in Science, titled Potential Biohazards of Recombinant DNA Molecules. This letter was coauthored by a pantheon of Nobel laureates and other scientific notables: Paul Berg, David Baltimore, Herbert W. Boyer, Stanley N. Cohen, Ronald W. Davis, David S. Hogness, Daniel Nathans, Richard Roblin, James D. Watson, Sherman Weissman, and Norton D. Zinder. This National Academy of Sciences committee stated the following:
"There is serious concern that some of these artificial recombinant DNA molecules could prove biologically hazardous. One potential hazard in current experiments derives from the need to use a bacterium like E. coli to clone the recombinant DNA molecules and to amplify their number. Strains of E. coli commonly reside in the human intestinal tract, and they are capable of exchanging genetic information with other types of bacteria, some of which are pathogenic to man. Thus, new DNA elements introduced into E. coli might possibly become widely disseminated among human, bacterial, plant, or animal populations with unpredictable effects."

The committee's recommendations included a request tothe director of the NIH to immediately consider the establishment of an advisory committee charged with (i) overseeing an experimental program to evaluate the potential biological and ecological hazards of the aforementioned types of recombinant DNA molecules; (ii) developing procedures to minimize the spread of such molecules within human and other populations; and (iii) devising guidelines for investigators working with potentially hazardous recombinant DNA molecules.
As chronicled elsewhere, the Recombinant DNA Advisory Committee (RAC) was established by the NIH in 1974 to provide recommendations to the NIH Director, as well as a public forum for discussion of the scientific, safety, and ethical issues related to basic and clinical research involving recombinant or synthetic nucleic acid molecules. In 2019, the NIH refocused the RAC into a role closer to its original mandate, which was to follow and advise on safety and ethical issues associated with emerging biotechnologies. Today, these emerging areas of research include technologies surrounding advances in recombinant or synthetic nucleic acid research. The RAC was renamed the Novel and Exceptional Technology and Research Advisory Committee (NExTRAC) to reflect its broader outlook.
Plasmids for Commercialization
The use of plasmids for commercialization has been the subject of numerous reviews written in various contexts. For this introductory blog, several points will be briefly mentioned. The first field that commercialization applied to was the biotechnology/biopharma industry. While Cohen returned to the laboratory in academia, venture capitalist Robert Swanson contacted Boyer to found Genetech in 1976. Boyer worked with Arthur Riggs and Keiichi Itakura from the Beckman Research Institute, and in 1977 the group became the first to successfully express a human gene in bacteria when they produced the hormone somatostatin. David Goeddel and Dennis Kleid were then added to the group and contributed to its success with synthetic human insulin in 1978. AMGen followed in 1980, and in the first three years, AMGen scientists attempted many things: creating organisms to extract oil from shale, growing chickens faster, making specialty chemicals, cloning luciferase (the light source for fireflies), and creating a process for producing indigo dye in E. coli—an achievement that garnered the prestigious cover of Science magazine in 1983.
The second context for commercialization using plasmids is gene therapy. The early years are discussed in a 2002 review in Trends in Biotechnology by Ferreira et al. titled Downstream processing of plasmid DNA for gene therapy and DNA vaccine applications. According to the reviewers, the use of viral vectors for gene delivery raised safety and regulatory concerns due to their toxicity and immunogenicity and the possible activation or deactivation of oncogenes or tumor-suppressor genes. "For these reasons, non-viral delivery systems became increasingly desirable, being the most attractive gene-transfer systems for commercial products. The number of approved gene-therapy protocols using plasmid-DNA-based delivery vectors has increased exponentially since 1995, representing approximately 25% of the ongoing gene-therapy trials."
The Path from Phage Biology to Plasmids for IVT mRNA Production
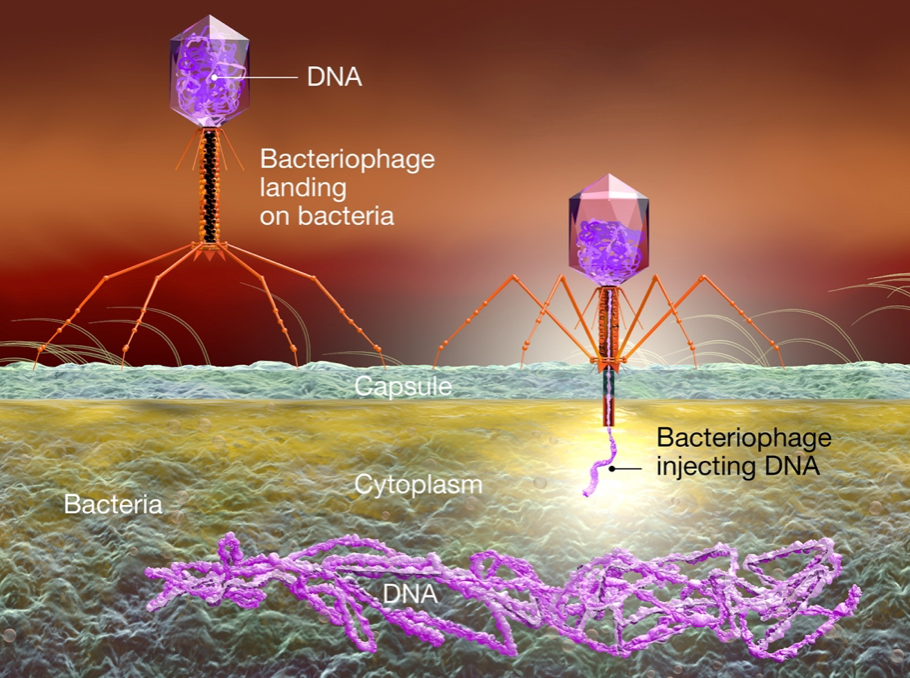
While the construction and use of plasmid DNA for the synthesis of mRNA by means of IVT is now common, the path to this technology began with early studies of the molecular biology of bacteriophages. These organisms, informally called phages, infect and replicate within bacteria and archaea, as illustrated here. The term was derived from "bacteria" and the Greek word "phagein," meaning "to devour." Bacteriophages are comprised of proteins that encapsulate a DNA or RNA genome.
Early studies of phage biology eventually led to molecular-level studies of how and when phages replicate in host organisms, especially bacteria. The phage designated as T7 was extensively investigated and, in 1973, Chamberlin and Ring reported the characterization of T7-specific RNA polymerase (RNAP). According to the researchers, the growth of T7 phage required the participation of two different DNA-dependent RNAPs. The bacterial RNAP was required in the initial stages of infection to transcribe the early regions of the T7 genome; however, this enzyme was restricted in its action to a region representing ~20% of the T7 DNA. Transcription of the remainder of the T7 genome required the protein product of T7 gene 1, which is a T7-specific RNAP.
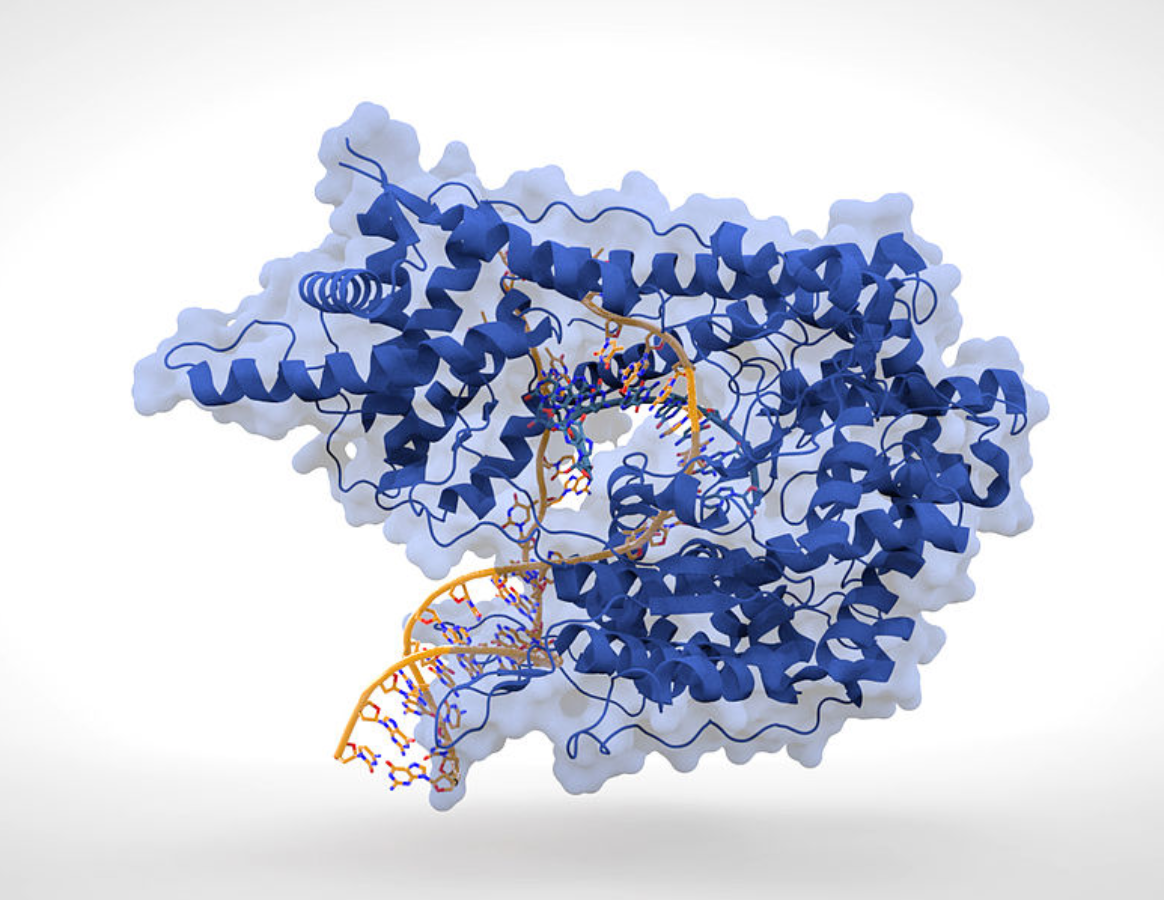
In light of this, Chamberlin and Ring investigated the general biochemical properties of both the enzymatic reaction carried out by T7 RNAP and the template specificity of the enzyme. The enzyme showed the same absolute requirements for DNA (nucleoside triphosphates) and a divalent metal ion similar to those found for bacterial RNA polymerases. The conditions for optimal RNA synthesis were, however, somewhat different. T7 RNAP initiates T7 RNA chains rapidly with GTP, and each enzyme molecule can initiate several T7 RNA chains in the course of the reaction. Hence, an efficient termination mechanism is present in the in vitro system. Based on various experimental results, Chamberlin and Ring suggested that "T7 RNAP requires a specific promoter site on DNA for effective transcription; this site is different from that used by bacterial RNAP."
A few years later, in 1979, Rosa published the DNA sequences of four promoters for the phage-specified T7 RNAP. All four regions contain the identical 23 bp sequence 5′ pTAATACGACTCACTATAGGGAGAOH, which includes the 6 bp of the transcribed region, as well as the 17 bp before it. This work, completed before the advent of current automated Sanger-based sequencing, used the original manual Maxam-Gilbert sequencing method.
As shown here, the position of the first transcribed nucleotide is commonly referred to as the +1 transcript nucleotide of that RNA, whereas the second transcribed nucleotide is referred to as the +2 transcript nucleotide, and so on. During transcription, the two DNA strands of the plasmid are melted to form a transcription bubble, and the bottom strand of the duplex (shown 3' to 5') is the template for transcription. For transcript nucleotides +3 and beyond, the template strand defines the identity of the transcribed nucleotides primarily through Watson-Crick base-pairing interactions. The nucleotide encoding the first RNA transcript nucleotide is therefore defined as the +1 nucleotide of the template. In the example shown here, the +1 transcript nucleotide is G, and the +1 template nucleotide is C, the +4 transcript nucleotide is A, and the +4 template nucleotide is T, and so forth.

IVT mRNA "Capping-ology"
As reviewed elsewhere, 5’-cap structures of the m7G(5')ppp(5')N(m)pN(m)p-type are present at the 5′ ends of nearly all eukaryotic cellular and viral mRNAs. A cap is added to cellular mRNA precursors and to the transcripts of viruses that replicate in the nucleus during the initial phases of transcription and before other processing events such as 3′-poly(A) addition or exon splicing. In the early days of synthesizing capped RNA transcripts in vitro for research, a phage RNAP bacteria, in the presence of all four NTPs and a cap dinucleotide such as m7GpppG, would be used to transcribe a DNA template.
The polymerase initiates transcription with nucleophilic attack by the 3’-OH of the G moiety in m7GpppG on the α-phosphate of the next templated NTP, resulting in the initial product m7GpppGpN. Suppression of the alternative GTP-initiated product pppGpN is achieved by using a five-to-ten-fold molar excess of m7GpppG to GTP in the transcription mixture.
In 1995, Pasquinelli et al. reported that phage RNAPs also utilize the 3’-OH of the 7-methylguanosine moiety of m7GpppG to initiate transcription, resulting in one-third to one-half of the products containing a "reverse cap," i.e., Gpppm7GpN. These authors demonstrated that when injected into Xenopus laevis nuclei, reverse-capped pre-U1 spliceosomal RNA transcripts are exported more slowly than natural transcripts. Similarly, cytoplasmic reverse-capped U1 RNAs fail to be imported into the nucleus.
In 2001, Stepinski et al. noted that although the presence of a cap on mRNA has a robust stimulatory effect on translation (reviewed by Shatkin, 1985), Pasquinelli et al. did not test the translation abilities of reverse-capped mRNAs. According to the researchers, based on what was known about recognition of the cap structure by eukaryotic translation initiation factor 4E (eIF4E), one would not expect reverse-capped mRNAs to be translated any more efficiently than uncapped RNAs. The presence of a significant proportion of reverse-capped RNAs in the products of in vitro transcription should decrease the overall translational activity of the RNA preparation.
Based on this reasoning, Stepinski et al. designed a cap analog that theoretically cannot be incorporated in the reverse orientation (shown here). They found that this "anti-reverse" cap analog (ARCA) is, in fact, incorporated exclusively in the normal orientation. Furthermore, it functions like the natural caps in its interactions with the translational machinery, resulting in mRNAs that are translationally more active.
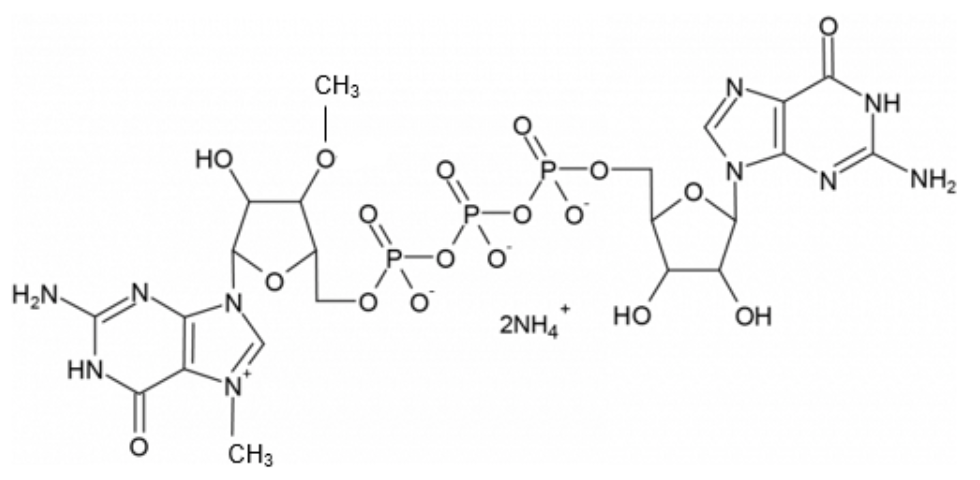
This ARCA analog reagent remained the state-of-the-art for co-transcriptional capping until 2018. Anton McCaffrey, others at TriLink, and additional collaborators reported that capping yields and IVT mRNA performance could be significantly improved using a new reagent called CleanCap® Reagent AG, shown here.
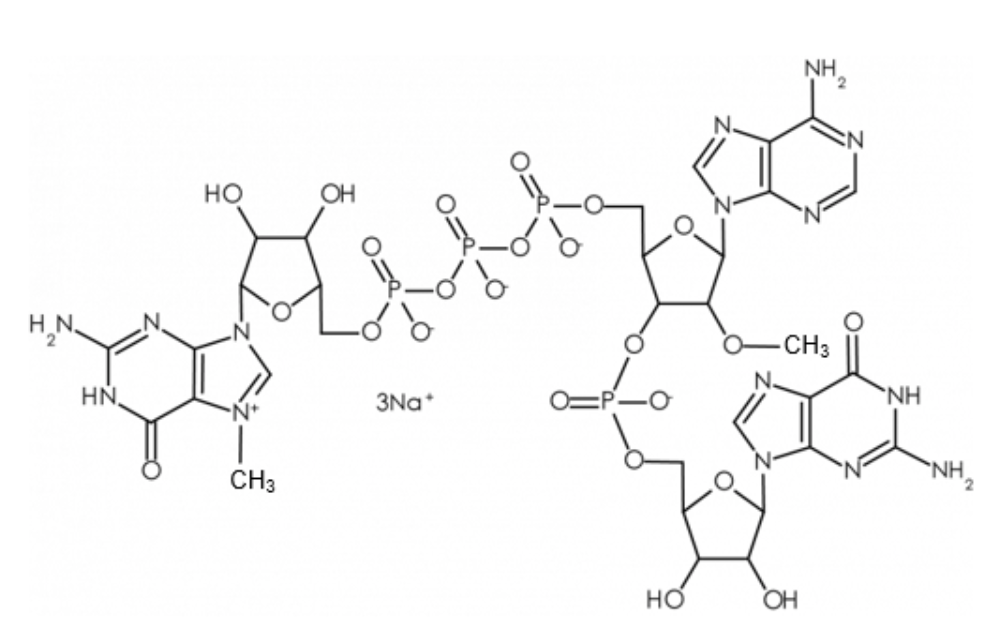
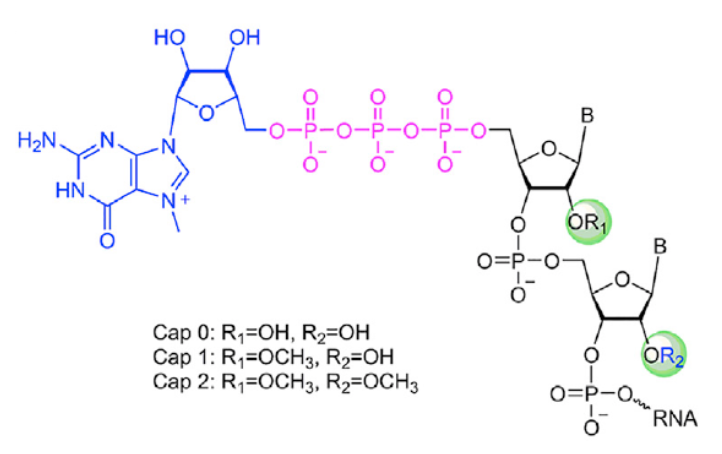
Readers interested in the design rationale and comparisons of CleanCap® Reagent AG with ARCA should consult the original publication for details. For this blog, and as shown here, it should be noted that CleanCap® Reagent AG yields a naturally occurring Cap1 structure. In higher eukaryotes, the 2'OH ribose position of the first cap-proximal nucleotide is methylated to form a Cap1 structure, and, in ~50% of transcripts, the second cap-proximal nucleotide is 2'O-methylated to form Cap2, shown here. In contrast, ARCA results in a Cap0 mRNA.
General Aspects of Plasmid DNA Manufacturing
As a consequence of intensive process development and engineering, a number of plasmid DNA manufacturing processes have been established and applied, as reviewed in several citations in a 2012 book chapter by Urthaler et al. titled Industrial Manufacturing of Plasmid-DNA Products for Gene Vaccination and Therapy. These authors further observe that this substantial amount of process development and engineering is also indicated by a large number of patents and patent applications in the field of plasmid upstream and downstream processing, for which they again cite several relevant reviews.
Urthaler et al. states that "despite the remarkably big variety of different production approaches, most plasmid DNA manufacturing processes are characterized by the following common features: (i) the bacterial host E. coli is used, and the corresponding E. coli replicons are utilized for propagation of host and plasmid in a bioreactor, (ii) alkaline lysis is applied to release the plasmid from the bacterial cells, and (iii) the plasmid is predominantly purified by chromatographic techniques and (ultra)filtration steps. As a representative example, a general plasmid DNA manufacturing flow chart is shown here, without the inclusion of in-process or final QC steps.
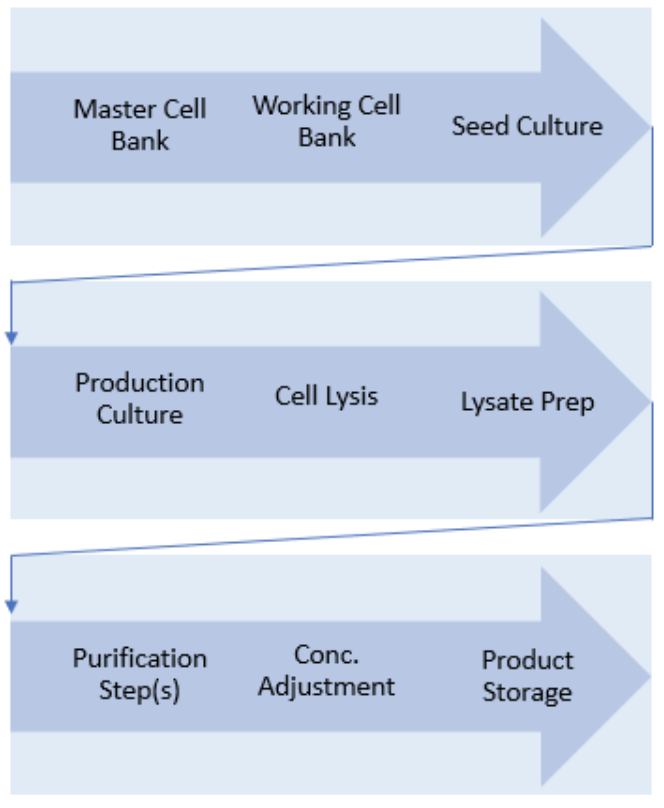
Thus, in contrast to the manufacturing of recombinant proteins that must include provisions for proper folding, crosslinking, and/or post-translational modification (i.e., glycosylation, etc.), advanced downstream processing concepts for industrially manufactured plasmid DNA are characterized by their generic applicability. This means that once established for a particular plasmid, a certain manufacturing process may be adapted to other plasmids with only a few or minor process changes.
Generic plasmid DNA processes offer a number of benefits, including a short project lead-time and reduced process development efforts for every new plasmid, as well as a relatively straightforward process validation timeline for mature projects. Another advantage of generic processes is that they are more easily transferable between different sites and facilities.
For the manufacturer's client, whether in academia or biopharma, it is critical to limit the number and degree of process changes during clinical development to reduce cost and time. In this context, the threat of pandemic diseases—as the world is now acutely aware of—has led to important considerations how plasmid DNA process engineering
could contribute to the supply of kilogram-scale amounts of plasmid DNA or IVT mRNA vaccines in a relatively short period of time.
To address this scale issue, technology co-operation between small- and large-scale contract manufacturing organizations (CMOs) is essential, as evidenced by the numerous news reports on COVID-19 vaccine development from 2020 through 2021. By applying a generic manufacturing process, technology transfer from one manufacturing company, site, or facility to another is considerably simplified, as product quality is not compromised and no major process changes are required.

Concluding Comments
In light of the brief, historically oriented perspectives discussed above, the Zone is struck by the multifaceted technical evolution of plasmids, from their initial discovery as agents for "genetic exchange" in 1952 to the current manufacturing of plasmid DNA for IVT mRNA production. During this nearly 50-year period, an amazing amount of basic science and applied R&D has been accomplished.
In view of the successful application of this combined technology for the manufacturing of both the Pfizer-BioNTech and Moderna IVT mRNA vaccines, plasmid manufacturing has played a vital role in making the world a safer place.
As a final comment, it is worth mentioning that in 2014, enzymatically produced circular DNA constructs replicated in vitro by rolling circle amplification were reported as alternatives to plasmids in DNA vaccines. While this "abiotic" approach that bypasses bacterial production of plasmids is conceivably applicable to non-plasmid-templated IVT mRNA production, the Zone could not find such publications.
Remember to be on alert for Part 2 of this series on plasmid manufacturing, which will provide additional perspectives highlighting key steps in perfecting these processes to guarantee the highest possible quality for translational research-to-clinic.
Your comments are welcomed, as usual.
Please feel free to share this blog with your colleagues or on social media.
PS: Remember to register for the June 15, 2021, TriLink mRNA Basics webinar: Getting Started with GMP mRNA Manufacturing.





