
Splicing to Save Memory
Alzheimer’s Disease (AD) is the most common form of dementia or progressive memory loss, affecting 5.8 million Americans. Approximately 90% of AD patients have the late-onset form of the disease, which presents with symptoms in the patient’s 60s. Late-onset AD is caused by a combination of environmental, lifestyle, and genetic risk factors, including single nucleotide polymorphisms (SNPs). However, a better understanding of the biological impact of these SNPs and how they could be treated remains a work in progress.
A SNP in exon 2 of the CD33 gene has been shown to reduce susceptibility for late-onset AD. This CD33 SNP affects alternative splicing in the CD33 mRNA. In the brain, CD33 is expressed primarily in microglial cells. Microglial activation is thought to prevent proper innate immune degradation of the beta amyloid plaques that accumulate to cause AD. The presence of this SNP leads to exon 2 exclusion, in turn reducing CD33 expression and, potentially, microglial activation. However, little is known about CD33 exon 2 splicing regulation.
Recently, a group of researchers at Pfizer published a study detailing the effects of this AD-associated SNP on the splicing and expression of CD33. Using a combination of genetic, functional genomic and proteomic approaches, these researchers identified two splicing regulatory proteins that influence CD33 exon 2 inclusion: SRSF1 and PTBP1.
To first demonstrate that the SNP’s effects on CD33 expression were due to splicing regulation, the authors compared mRNA (containing or lacking exon 2) and protein expression after transfecting HeLa cells with minigenes (with introns) or cDNA constructs (without introns). The SNP genotype only affected expression when introns were present. Importantly, the authors used TriLink’s EGFP mRNA (L-6101) to generate a genome-edited cell line lacking CD33 expression and confirmed the specificity of their FACS protein expression assays throughout the paper.
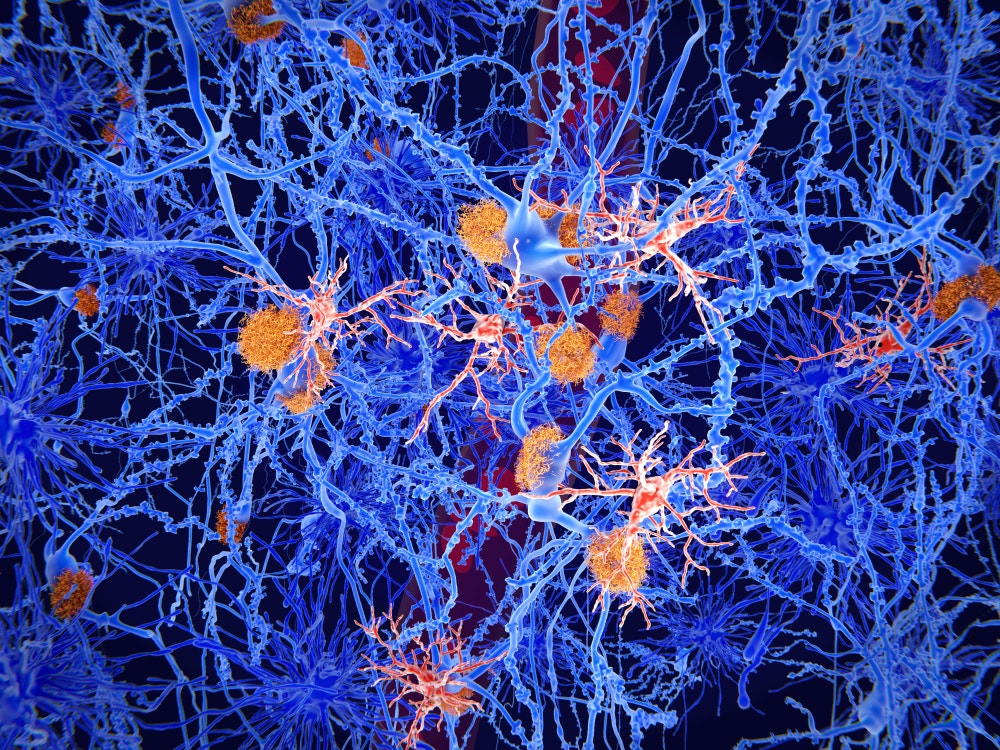
Figure 1: Microglia cells (red) play an important role in the pathogenesis of Alzheimer's disease. Microglia are specialised macrophages that restrain the accumulation of amyloid (orange plaques).
With complementary siRNA knockdown and overexpression studies, the authors showed that SRSF1 binding enhanced exon 2 inclusion. Interestingly, SRSF1 binding at the 3’ end of exon 2, but not at the 5’ end near the SNP, affected exon 2 inclusion. Through SILAC mass spectrometry and subsequent experiments, the researchers showed that PTBP1 binds much more efficiently at the higher AD risk allele to promote exon 2 inclusion. Thus, SRSF1 and PTBP1 both function as splicing enhancers to promote exon 2 inclusion and full length CD33 mRNA expression, leading to higher CD33 surface protein expression.
Although this paper focuses on the details of splicing regulation and mRNA-protein binding events, the authors did not demonstrate effects of SRSF1 or PTPB1 on CD33 protein function or microglial activation. Yet, this work supports future research into using ASOs or small molecules to block SRSF1 or PTBP1 binding, as this not only results in the exclusion of exon 2 in CD33 and the inhibition of microglial activation, but also potentially reduces the risk of late-onset AD.
Featured products: CleanCap® EGFP mRNA (L-7601); CleanCap® EGFP mRNA (5moU) (L-7201)