- 1,000 Genomes Project is Big
- 10,000 Genomes Project is Bigger
- 100,000 Genomes Project is Biggest—so Far
- Will 1,000,000 Genomes be Next?
This blog on genomics projects going democratic has—rest assured—nothing to do with US presidential election politics that are already receiving (too much) 24/7 coverage—but rather genomics going from singular to pluralistic. Let me frame this revolutionary change another way to clarify: the much heralded sequencing of “the human genome” (singular) announced in 2001—by competing public and private initiatives—used mixtures of DNA from multiple donors, i.e. “the genome” was actually “the genomes,” all of which are different—in some way. These differences are what make each of us genetically unique. Consequently—and enabled by ever faster and cheaper DNA sequencing—there are increasingly large projects aimed at identifying these genetic variations (aka genotypes or polymorphisms) for association with health or disease status (aka phenotypes). To me, this fundamentally important trending science is definitely blogworthy.
 Populations are comprised of genetically unique individuals. Taken from my Wakulla.com.
Populations are comprised of genetically unique individuals. Taken from my Wakulla.com.
Genotyping-Phenotyping “101”
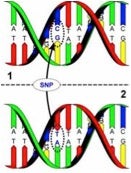 Individuals 1 and 2 have a SNP in this particular stretch of their respective genomes. Taken from broadinstitute.org.
Individuals 1 and 2 have a SNP in this particular stretch of their respective genomes. Taken from broadinstitute.org.
Geneticists have been well aware that sequencing DNA from different individuals leads largely to regions of sequence identity between all or some individuals, and other types of sequence differences. Roughly speaking, we are about 99% identical and 1% different in terms of DNA sequence. It has also been known that these sequence differences would be either stretches of identical DNA with an occasional single-base difference—called a single nucleotide polymorphism (SNP)—or other multiple-base sequences of DNA that were inserted or deleted (indel).
These SNPs, indels, and other genetic variant classifications are what actually underlies individuals having different physical traits—such as eye, hair, and skin colors—or different genetic predispositions to medical conditions during life, or odds of being born with a genetic disease. Consequently, sequencing DNA from more and more individuals is needed to generate enough data to analyze and then—hopefully—understand its theoretical and practical significance. Like it or not, “heavy duty” statistics—not my forte—comes into play, as indicated by this website on statistical genetics.
Having said this, you can now appreciate that details related to how many more individual DNA samples are sequenced, which populations of individuals are studied, and—importantly—what’s done with the data are all open-ended topics of debate. In addition, some opine concerns for ethical issues and individual privacy.
Although I’m quite interested in these latter debates, the main objective is to inform you about two recently published population-based studies involving a very large numbers of individuals (i.e. thousands and tens of thousands) and an ongoing mammoth analysis of a hundred thousand individuals. Hence this blog’s headline of “big, bigger, biggest”—with a byline hint of bigger than biggest yet to come.
1,000 Genomes Project is Big
Two papers published in the October 1, 2015 issue of venerable Nature magazine report on the final phase of the 1000 Genomes Project. Begun in 2008, the project has developed an open—i.e. publically available—resource that now includes the genomic data for 2,504 individuals from 26 global populations, all analyzed using whole-genome sequencing, exome (e.g. mRNA in the form of cDNA) sequencing and microarray genotyping approaches. The main report from the consortium of 843 (!) collaborators describes key features of all 88 million genetic variants identified. These include 84.7 million SNPs, 3.6 million short indels, and 60,000 structural variants.
These polymorphisms are displayed in several graphs shown below. While these are better viewed in full size and further explained in the aforementioned open publication, it can be seen in graph (a) that, for example, US-located individuals (ASW) harbor significantly different proportions of variants than Great Britain-located individuals (GBR). Furthermore, graph (b) reveals that ASW vs. GBR harbor more variant sites per genome (~4.7 vs. ~4.1 per million bases, respectively). Simply put, there are genetic variations among different geographical populations.
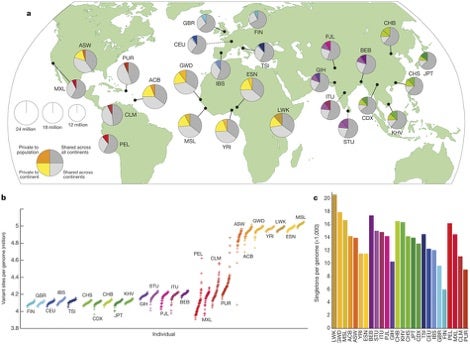 (a) The area of each pie is proportional to the number of polymorphisms within a population. Pies are divided into four slices, representing variants private to a population (darker color unique to population), private to a continental area (lighter color shared across continental group), shared across continental areas (light grey), and shared across all continents (dark grey). Dashed lines indicate populations sampled outside of their ancestral continental region. (b) The number of variant sites per genome. (c) The average number of singletons per genome.
(a) The area of each pie is proportional to the number of polymorphisms within a population. Pies are divided into four slices, representing variants private to a population (darker color unique to population), private to a continental area (lighter color shared across continental group), shared across continental areas (light grey), and shared across all continents (dark grey). Dashed lines indicate populations sampled outside of their ancestral continental region. (b) The number of variant sites per genome. (c) The average number of singletons per genome.
As demonstrated by the data above, these population-specific genotype-phenotype databases need to be considered for various actionable applications such as DNA-based forensics, diagnostics, or therapeutics. Other applications range from hunts for the genetic roots of human illness to analyses of population genetics and evolutionary history.
The accompanying Nature editorial entitled Variety of Life offered this take home message: “although most common genetic variants are shared across populations, rarer variants are often restricted to closely related groups. Many more rare variants are still to be identified.” I agree, and have observed over all of the years since genome sequencing was envisaged, that the need for faster and more inexpensive sequencing methodology continually begot even faster and yet cheaper methodology, almost like a self-fulfilling fantasy. But it’s really happening, and I digress…so let’s move on to the next big thing—to quote Steve Jobs.
10,000 Genomes Project is Bigger
“Size does matter” for genome projects aimed at identifying less common or rare DNA sequence variants due to the need to sequence more individuals. This is akin to finding a smaller needle in a larger haystack, relative to identifying so-called common variants that, by definition, occur more frequently in a given population. Consequently, whole genome sequencing of 10,000 individuals is statistically needed in the eponymously named UK10K project that intends to enable researchers in the UK and elsewhere to “better understand the link between low-frequency and rare genetic changes, and human disease caused by harmful changes to the proteins the body makes.” Incidentally, UK10K defines low-frequency as 1-5% minor allele frequency (MAF) and rare as <1% MAF.
Another technical factor worth recognizing with regard to all sequencing studies is so-called sequence “read depth” (aka depth of coverage), which takes into account the average number of times stretches of sequences are detected or read. The more times they are read the better, because greater read depth leads to higher statistical significance of sequence variants actually being real as opposed to a sequencing artifact.
In the aforementioned Oct 1, 2015 issue of Nature, a consortium of 321 (!) collaborators reports sequencing results from low read depth (7×) of whole genomes or high read depth (80×) of exomes of nearly 10,000 individuals from population-based and disease collections. Remarkably—to me—sequencing for this massive study began in late 2010, so if there were only about 4 years of data collection, genomes and exons from ~2,500 individuals were sequenced per year or ~7 per day, 365 days per year—that’s fast.
From cohorts of different populations having extensive medical records, the study found >24 million novel sequence variants and identified novel alleles associated with levels of triglycerides (APOB), adiponectin (ADIPOQ) and low-density lipoprotein cholesterol (LDLR and RGAG1). All of these levels are related to probability of cardiovascular diseases (CVDs).
Why focus on CVDs? The answer is clear from the following facts that I found provided by the World Health Organization, with the last point italicized by me because of its relevance to the aforementioned findings of UK10K:
- More people die annually from CVDs than from any other cause.
- An estimated 17.5 million people died from CVDs in 2012, representing 31% of all global deaths. An estimated 7.4 million were due to coronary heart disease and 6.7 million were due to stroke.
- Over three quarters of CVD deaths take place in low- and middle-income countries.
- Most cardiovascular diseases can be prevented by addressing behavioural risk factors such as tobacco use, unhealthy diet and obesity, physical inactivity and harmful use of alcohol using population-wide strategies.
- People with cardiovascular disease or who are at high cardiovascular risk (due to the presence of one or more risk factors such as hypertension, diabetes, hyperlipidaemia or already established disease) need early detection and management using counselling and medicines, as appropriate.
 Jerry’s advice: “love your heart—it’s the only one you have.” Taken from Punnett.blogspot.com.
Jerry’s advice: “love your heart—it’s the only one you have.” Taken from Punnett.blogspot.com.
Although many hundreds of genes that are involved in causing disease have already been identified, it is believed that many more remain to be discovered. The UK10K project aims to help uncover them by studying the genetic code of 10,000 people in much finer detail than ever before
100,000 Genomes Project is Biggest—so Far
I’m impressed by UK stepping up, if you will, to take the lead on executing its UK10K genomes project, and I’m super impressed by its pushing even further to transform the UK National Health Service (NHS)—akin to the US Medicare/Medicaid system—via the 100,000 Genomes Project. The stated intent is to create “a new genomic medicine service for the NHS—transforming the way people are cared for. Patients may be offered a diagnosis where there wasn’t one before. In time, there is the potential of new and more effective treatments.”
The 100,000 Genomes Project website is definitely worth visiting. I suggest watching the prominently displayed video that nicely provides commentary from several people involved in this project of how this landmark effort will transform the NHS for the better. It gave me an appreciation of real people advocating real change.
This website homepage also has a link to a real-time progress count of the number of genomes sequenced thus far. As of November 2nd, 5,630 genomes had been sequenced. To put this number in perspective, I found a wiki stating that, as of August1, 2014 the project had passed the 100 genome mark, with the aim of reaching 1,000 by the end of 2014, 10,000 by the end of 2015 and 100,000 by 2017. It also stated that the project is initially focusing on patients with rare diseases and their families, as well as patients with some common types of cancer.
Will 1,000,000 Genomes be Next?
Seemingly not to be outdone by the UK, President Obama announced in January 2015, and NIH has posted, plans for what is called the Personalized Medicine Initiative (PMI). Although President Obama’s statements about this initiative generated—as expected—plenty of press and social media buzz that are more soundbites and attention-grabbers than accurate facts, I strongly advise interested seekers-of-facts to peruse the NIH website, as I did to learn factual details regarding PMI.
 Left-handed President Obama pointing to right-handed DNA helix. Taken from America.Aljazeera.com
Left-handed President Obama pointing to right-handed DNA helix. Taken from America.Aljazeera.com
Speaking of details, there is a lengthy (108 page) downloadable pdf dated September 17, 2015 by a Working Group to the Advisory Committee to the Director, NIH. It covers all facets of PMI including, sections on the following:
- The “promise of precision medicine”
- Assembling the cohort of 1,000,000 or more PMI volunteers
- Engagement of PMI “participant as partners”
- Acquiring research data
- Biobanking specimens
- Privacy, misuse of information, and security
- Participant access to data
Despite the great job of outlining all of these—and many more—complex operational aspects, I was disappointed by the following description of what methods would be employed to obtain laboratory data for biospecimens: “Genomics, proteomics, metabolites, cell-free-DNA, single-single cell studies, infectious exposures, standard clinical chemistries, histopathology.” We’ll have to wait to learn how many genomes are ultimately sequenced. Also, I looked for a projected timeline for completion and overall total budget, but was unsuccessful.
I hope this is not like Nancy Pelosi’s infamous quote that "[w> e have to pass the [health care> bill so you can find out what is in it,"
Postscript
After writing this blog, I saw this tweet by Broad Genomics (@BroadGenomics) which—if true—blows my mind because of the claimed throughput achieved by whatever factory-like workflow and instrumentation they must have:
“Back of the napkin calculation: Last month we produced a 30× human genome about every 10 minutes.”
 Taken from a tweet by the Broad Genomics (@BroadGenomics).
Taken from a tweet by the Broad Genomics (@BroadGenomics).




