- Like it or not, and for better or worse, next-generation sequencing is revealing that you and your bacterial microbiome have an inextricable biological relationship.
- “RePOOPulating” the gut: a clinical study of “synthetic stool” as a better alternative to fecal transplant.
- Microbiome movies, fungus too, and much more.
What’s in your microbiome? Why does it matter?
Writing this blog was inspired by reading Michael Pollan’s recent article in The NY Times Magazine entitled “Some of my best friends are bacteria,” which is an engaging story about Michael having his microbiome sequenced as part of the American Gut project. He notes at the beginning that each of us has several hundred microbial species with whom we share our body, and that these bacteria—numbering ~100 trillion—are living (and dying) right now on the surface of our skin, mouth, and intestine “where the largest contingent of them will be found, a pound or two of microbes together forming a vast, largely uncharted interior wilderness that scientists are just beginning to map.” The sheer numbers of these microbes, he adds, makes us only ~10% human—for every human cell there are ~10 resident microbes—most being “harmless freeloaders” or “favor traders” (i.e. symbiotic), and only a tiny number of pathogens. Furthermore, “[t>
his humbling new way of thinking about the self has large implications for human and microbial health, which turn out to be inextricably linked.”
If you want to know what’s in your gut, you can participate in this world wide study by registering at www.americangut.org. According to the website, you’ll be asked to ‘fill out a diet & lifestyle questionnaire (online) with such things as age, gender, weight, have you taken antibiotics lately, any conditions we should know about it, and so on.’ Anyone over the age of 3 months can participate in the study, and you will have the opportunity to provide a stool, tongue and/or palm sample via an at-home sample kit.
Microbes and Your Health
As more microbiome data is obtained from the American Gut project and analogous studies, correlations with each person’s health status can be made. Having the “wrong” kind of microbes may be associated with predisposition to obesity or certain chronic diseases, for example. Such information may then be used to prescribe dietary or other sources supplemental probiotics. An extreme outcome could involve “fecal transplants” (i.e., fecal microbiota transplantation or fecal bacteriotherapy) wherein a healthy person’s fecal microbiota are installed into a sick person’s gut.
My PubMed searches of these terms led to a number of publications, such as that by Christopher. R. Kelly et al. at Women and Infant's Hospital, Brown University Alpert School of Medicine, who reported promising results for relapsing Clostridium difficile infection in a small study of 26 patients. Coincidentally, on June 18th there was a report that the US Food & Drug Administration (FDA) is “dropping plans to tightly control” fecal transplants that are “becoming increasingly popular for treating people stricken by life-threatening infections of the digestive system.” This report went on to say that, in the FDA’s view, “such a treatment should only be given to patients who have exhausted other treatment options and who have given consent and been informed that it is an experimental procedure with risks.”
RePOOPulating the gut: a better alternative?
Among the concerns for fecal transplants are pathogen transmission, patient acceptance and inability to standardize the treatment regime, states Elaine O. Petrof and her colleagues in a study entitled Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut, recently published in Microbiome (2013). This online, open-access article is well worth a quick read if you’re interested in the experimental details, which take advantage of next-generation sequencing as a key method for quite sophisticated identification and analysis of microbes. In brief, a stool substitute preparation—made from 33 purified intestinal bacterial cultures derived from a single healthy donor—was used to treat two patients who had failed at least three courses of metronidazole or vancomycin. Pre-treatment and post-treatment stool samples were analyzed by 16S rRNA sequencing using the Ion Torrent platform. Both patients were infected with a hyper virulent C. difficile strain, but following treatment each reverted to their normal bowel pattern within 2-3 days and remained symptom-free at 6 months. Analysis demonstrated that rRNA sequences found in the stool substitute were rare in the pre-treatment stool samples, but constituted >25% of the sequences 6 months after treatment.
Microbiome Movies
Relatively low-cost deep-sequencing technology has enabled novel studies aimed at obtaining, in effect, “moving pictures of the human microbiome,” which is the attention-grabbing title of a study by J. Gregory Caparaso et al. in Genome Biology. This landmark publication in 2011 presented the largest human microbiota time-series analysis to date, covering two individuals each at 4 body sites over 396 time-points. One male and one female each provided gut (feces), mouth, left palm, and right palm samples over 15-mo and 6-mo periods, respectively, Variable regions of 16S ribosomal RNA (rRNA) in each sample were amplified by PCR and sequenced on an Illumina instrument. The following stated results and conclusions may surprise you, as they did me:
“We find that despite stable differences between body sites and individuals, there is pronounced variability in an individual’s microbiota across months, weeks and even days. Additionally, only a small fraction of the total taxa found within a single body site appear to be present across all time points, suggesting that no core temporal microbiome exists at high abundance (although some microbes may be present but drop below the detection threshold). Many more taxa appear to be persistent but non-permanent community members.”
“Because of the immense subject-to-subject variability in the microbiome, studies examining temporal variability, which give a view of dynamics beyond the static pictures previously available, have the potential to transform our understanding of what is ‘normal’ in the human body, and, perhaps, to develop predictive models for the effects of clinical interventions.”
If you’re questioning whether the above study of only two individuals—albeit for many time points—reflects a wider population, rest assured that it apparently does. The recently completed 5-yr Human Microbiome Project (HMP) launched in 2008 involved 242 volunteers, more than 5,000 samples were collected from tissues from 15 (men) and 18 (women) at body sites such as mouth, nose, skin, lower intestine (stool) and vagina. According to the HMP wiki site, this project’s discoveries include:
- Microbes contribute more genes responsible for human survival than humans' own genes. It is estimated that bacterial protein-coding genes are 360 times more abundant than human genes.
- Microbial metabolic activities—for example, digestion of fats—are not always provided by the same bacterial species. The presence of the activities seems to matter more.
- Components of the human microbiome change over time, affected by a patient disease state and medication. However, the microbiome eventually returns to a state of equilibrium, even though the composition of bacterial types has changed.
Consequently, defining what is “normal” and whether a “core” community of microbes exists is quite complicated, and will likely remain a topic of great interest and debate. In this regard, I found an overview by Dirk Gevers et al. well worth reading. Here’s one section of text that reiterates what I think are important points, and elaborates upon the above second bullet point about microbial metabolic activities, i.e. functional core:
“A potentially more universal ‘core’ human microbiome emerged during the consideration of microbial genes and pathways carried throughout communities' metagenomes. While microbial organisms varied among subjects as described above, metabolic pathways necessary for human-associated microbial life were consistently present, forming a functional ‘core’ to the microbiome at all body sites. Although the pathways and processes of this core were consistent, the particular genes that implemented them again varied. Microbial sugar utilization, for example, was enriched for metabolism of simple sugars in the oral cavity, complex carbohydrates in the gut, and glycogen/peptidoglycan degradation in the vaginal microbiome. The healthy microbiome may thus achieve a consistent balance of function and metabolism that is maintained in health, but with fine-grained details personalized by genetics, early life events, environmental factors such as diet, and a lifetime of pharmaceutical and immunological exposures.”
Lest you think that such data are collections of facts devoid of utility, think again after reading the following excerpts from the abstract of a publication by Fredrik H. Karlsson et al. in Nature 2013 entitled Gut metagenome in European women with normal, impaired and diabetic glucose control.
“Type 2 diabetes (T2D) is a result of complex gene–environment interactions, and several risk factors have been identified, including age, family history, diet, sedentary lifestyle and obesity. Statistical models that combine known risk factors for T2D can partly identify individuals at high risk of developing the disease. However, these studies have so far indicated that human genetics contributes little to the models, whereas socio-demographic and environmental factors have greater influence.…Here we use shotgun sequencing to characterize the fecal metagenome of 145 European women with normal, impaired or diabetic glucose control. We observe compositional and functional alterations in the metagenomes of women with T2D, and develop a mathematical model based on metagenomic profiles that identified T2D with high accuracy.”
Microbiomes in Homes and Hospitals
Mapping the great indoors—an engaging NY Times article by Peter A. Smith—tells about microbiologists who are using sequencing to “take a census” of what lives in our homes with us and how we “colonize” spaces with other species — viruses, bacteria, microbes. One group has sampled the “microbial wildlife” in 1,400 homes across the USA in a project called The Wild Life of Our Home, which relies on volunteers to swab pillowcases, cutting boards and doorjambs, then send samples in for analysis. Although data are still being analyzed, an earlier study of 40 homes in 9 locations around the Raleigh-Durham area of North Carolina has been published by Robert R. Dunn et al. in PLoS ONE entitled Home Life: Factors Structuring the Bacterial Diversity Found within and between Homes. Among their findings were the following.
“[E>
ach of the sampled locations harbored bacterial communities that were distinct from one another” [and that>
“the presence of dogs had a significant effect on bacterial community composition in multiple locations within homes as the homes occupied by dogs harbored more diverse communities and higher relative abundances of dog-associated bacterial taxa. Furthermore, we found a significant correlation between the types of bacteria deposited on surfaces outside the home and those found inside the home, highlighting that microbes from outside the home can have a direct effect on the microbial communities living on surfaces within our homes.”
 Your dirty dog? (Bing Images)
Your dirty dog? (Bing Images)
Some of you might be wondering what microbes your dog is depositing in your home, while others might be asking about cats. I haven’t a clue about your dog, but cats weren’t directly assessed in this study. Thirteen of the houses had only dogs as pets and the influence of dogs is likely to be greater than cats because of their large size and need to go outdoors. Only three houses had cats but not dogs (three houses had both), and the investigators judged this to be too small a sample size for cats.
The figure shown below taken from the aforementioned Dunn et al. publication tracks 9 sites sampled with 5 sources of microbes. The authors note that “[t>
hese results show changes in the relative importance of individual sources across sites, not comparisons across sources within sites. For example, these results show that soil is a more important source of bacteria on door trims than on cutting boards, but these results cannot be used to directly compare the relative importance of soil versus leaves as sources of bacteria at individual locations.” By the same token, the results show that human skin is a more important source of bacteria on toilet seats than on cutting boards.
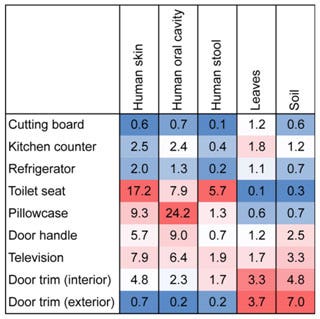 Source tracking analysis showing relative proportion of bacteria at each sampling site associated with given sources. Values represent median percentages. Warmer colors indicate greater influences of particular sources across the sites (Robert R. Dunn et al. PLoS ONE 2013).
Source tracking analysis showing relative proportion of bacteria at each sampling site associated with given sources. Values represent median percentages. Warmer colors indicate greater influences of particular sources across the sites (Robert R. Dunn et al. PLoS ONE 2013).
Microbial fingerprints
Roughly 1.7 million hospital-associated infections are reported each year in the USA, and the pathogens that cause them must come from somewhere. Beth Mole (too bad she wasn’t named Millie!) in Nature-News 2013 notes that patients leave a microbial mark on hospitals. This attention getting punch line refers to preliminary findings from the Hospital Microbiome Project that, according to its website, “aims to collect microbial samples from surfaces, air, staff, and patients from the University of Chicago's new hospital pavilion in order to better understand the factors that influence bacterial population development in healthcare environments.”
According to Beth Mole, “…even in areas with long-term inhabitants, [Jack>
Gilbert’s team has found no lingering pathogens. ‘Over the first four months of observations, we’ve seen nothing that concerns us,’ he says.” However, “Gilbert and his team found significant differences between microbial communities in individual hospital rooms. Patients who stayed for only short periods, such as those undergoing elective surgery, had a transient influence on their rooms’ microbial communities; after cleaning, the rooms reverted to a pre-patient state.” In contrast, and of concern, ‘[m>
icrobes from long-term patients—including people with cancer or those who had received organ transplants—had time to settle into the rooms. The patients' microbial fingerprints lingered after they checked out of the hospital, even after their rooms were cleaned." But even in areas with long-term inhabitants, Gilbert’s team has found no lingering pathogens. “Over the first four months of observations, we’ve seen nothing that concerns us,” he says.”
Thinking Big: The Global Microbiome
If you now muse about greatly expanding the scope of the aforementioned studies to a global scale, you’d be thinking about something that has already been proposed. The Earth Microbiome Project is a “massively multidisciplinary effort to analyze microbial communities across the globe” that proposes to “characterize the Earth by environmental parameter space into different biomes and then explore these using samples currently available from researchers across the globe.” The project intends to “analyze 200,000 samples from these communities using metagenomics, metatranscriptomics and amplicon sequencing to produce a global Gene Atlas describing protein space, environmental metabolic models for each biome, approximately 500,000 reconstructed microbial genomes, a global metabolic model, and a data-analysis portal for visualization of all information.” We’ll all have to stay tuned on this rather ambitious project to learn what is found and, more importantly, what are the conclusions—and whether time-dependent variability is accounted for.
By the way, if you’re wondering about the total global microbiome, Addy Pross’ book entitled What is Life? How Chemistry Becomes Biology states that the earth’s bacterial biomass is estimated to be 2 × 1014 tons, which is sufficient to cover the earth’s land surface to a depth of 1.5 meters!
Microbiome-mania & Toilet-phobia
There seems to be a surge in extending microbiome analysis to many other contexts. Some of these findings are surprising—and prompting jokes—or might scare you about microorganisms present in various places or on things that we come into contact with, whether we know it or not.
Initial, a UK-based provider of hygiene services to businesses and organizations, announced in an April 2013 press release, its research that “…lifts the lid on the grimy state of Britain’s office kitchens.” Being a provider of cleaning services, Initial was presumably quite pleased to inform the public about the following findings.
“Swab testing of a sample of communal workplace kitchens showed that 75% of work surfaces were home to more bacteria than an average feminine sanitary bin. Half also harboured dangerously high levels of coliforms, bacteria present in feces, which can lead to outbreaks of gastrointestinal disease. Over a quarter of the draining boards tested registered more than four times the level of coliforms considered to be safe.”
“The handles of shared fridge-freezers were also shown to be bacteria-rife, with a third carrying high levels of coliforms, whilst 30% of shared microwaves were also shown to be contaminated around the handles and buttons.”
“Tea drinkers were no more hygienic, with over 40% of kettle handles found to be carrying high levels of bacteria, and significantly exceeding the bacteria levels on toilet doors. Also, tested were cupboard, dishwasher and waste bin handles, with the cleanest appliance in the kitchen proving to be the water cooler.”
Speaking of toilets, they seem to be in vogue in metabolome studies. For example, Emma Innes reported online that women's handbags contain more bacteria than the average toilet seat. The dirtiest item in an average handbag is hand cream—it carries more bacteria than the average toilet seat. Leather handbags carry the most bacteria because the spongy texture provides “perfect growing conditions.” An online article by Harold Maass noted that British researchers found that the average barbecue grill in the UK has more than twice as many germs as the typical toilet seat. Maybe someone has already invented a disposable barbeque grill cover similar to what’s used to cover toilet seats, but obviously nonflammable.
 Sanitor Mfg Co. began producing toilet seat covers and dispensers in the USA in 1931. Seems they are working as your handbag and your barbeque may both harbor more germs than a toilet seat.
Sanitor Mfg Co. began producing toilet seat covers and dispensers in the USA in 1931. Seems they are working as your handbag and your barbeque may both harbor more germs than a toilet seat.
Although humorously entitled as Lifting the lid on toilet plume aerosol, this review article recently published the American Journal of Infection Control examines the evidence regarding toilet plume bioaerosol generation and infectious disease transmission. Here’s a quote of the results of this review of existing literature. “The studies demonstrate that potentially infectious aerosols may be produced in substantial quantities during flushing. Aerosolization can continue through multiple flushes to expose subsequent toilet users. Some of the aerosols desiccate to become droplet nuclei and remain adrift in the air currents. However, no studies have yet clearly demonstrated or refuted toilet plume-related disease transmission, and the significance of the risk remains largely uncharacterized.” Like many investigations, the stated conclusions call for more data: “[a>
dditional research in multiple areas is warranted to assess the risks posed by toilet plume, especially within health care facilities.
By the way, the internet has various references to a 6-foot diameter (dare I say) zone wherein toilet aerosol may spread, so keep your toothbrush at a safe distance or consider purchasing a toothbrush sanitizer—of which there are many—after doing your homework on which devices have been validated, and against what, as I did by searching PubMed for “toothbrush sanitizer” etc.
Mycobiome: Fungus In, On and Among Us
Oh…let’s not forget about fungus—a member of a large group of microorganisms that includes yeasts and molds, as well as mushrooms. These organisms are classified as a kingdom, Fungi, and the discipline of biology devoted to the study of fungi is known as mycology. My PubMed search of “mycobiome” gave only 10 hits, which is far less than ~4,000 found for “microbiome.” The earliest mycobiome publication was entitled “Characterization of the oral fungal microbiome (mycobiome) in healthy individuals” by Mahmoud A. Ghannoum et al. (PLoS Pathogens 2010). This study—which used pyrosequencing to characterize fungi present in the oral cavity of 20 healthy individuals—revealed the "basal" oral mycobiome profile of the enrolled individuals and showed that across all the samples studied, the oral cavity contained 74 culturable and 11 non-culturable fungal genera. The oral mycobiome of at least 20% of the enrolled individuals included the four most common pathogenic fungi—Candida (present in 75% of the cohort; mostly C. albicans), Aspergillus (35%), Fusarium (30%), and Cryptococcus (20%). The authors said that “[i>
t is possible that the pathogenicity of these fungi is controlled in healthy individuals by other fungi in the oral mycobiome, as well as a functional immune system.”
 Candida albicans (Wikipedia via Bing Images)
Candida albicans (Wikipedia via Bing Images)
More recently, Heidi H. Kong and coworkers in Nature 2013 published a report entitled Topographic diversity of fungal and bacterial communities in human skin. This study involved 10 healthy individuals and used sequencing to analyze fungal and bacterial communities sampled from 14 skin sites that included face, chest, arms, ears, nostrils, head, and feet. Some salient points taken from the abstract are as follows.
“Eleven core-body and arm sites were dominated by fungi of the genus Malassezia, with only species-level classifications revealing fungal-community composition differences between sites. By contrast, three foot sites—plantar heel, toenail and toe web—showed high fungal diversity. Concurrent analysis of bacterial and fungal communities demonstrated that physiologic attributes and topography of skin differentially shape these two microbial communities. These results provide a framework for future investigation of the contribution of interactions between pathogenic and commensal fungal and bacterial communities to the maintenance of human health and to disease pathogenesis.”
Lastly, Who was First?
Who was “first-to-publish” and what was envisaged have always been of interest to me. In researching this posting, I became curious about who introduced the term “microbiome” to generally describe concepts of the type represented in the various aforementioned articles. It’s not easy to be find or establish “first-ever” publications, so I took the easy way out by doing a PubMed search wherein “microbiome” is in the title and/or abstract. Of the ~1,400 articles found, the earliest was entitled New technologies, human-microbe interactions, and the search for previously unrecognized pathogens by David A. Relman at Stanford University, and appeared in Journal of Infectious Diseases in 2002. The following conclusion from his abstract is quite prescient, and I’m amazed by how far and fast microbiome science has progressed since then.
“The development and clinical application of molecular methods have led to the discovery of novel members of the endogenous normal flora as well as putative disease agents. Current challenges include the establishment of criteria for disease causation and further characterization of the human microbiome during states of health. These challenges and the goal of understanding microbial contributions to inflammatory disease may be addressed effectively through the thoughtful integration of modern technologies and clinical insight.”
As always, your comments are welcome—especially if you know of a particularly interesting “microbiome” report that you’d like to share with me and other readers of this blog.
Postscript
After writing this blog, GenomeWeb reported on August 8th that The Wellcome Trust has awarded $2 million to fund Lindsay Hall, a researcher at the University of East Anglia and the Institute of Food Research, who will seek to find out how bacteria that are beneficial to humans help protect against diseases in the early phases of life, using high-throughput sequencing tools to find out more about the microbial communities that colonize the human body soon after birth. "We are planning to use 16S rRNA-based microbiota community analysis, metagenome, and whole genome sequencing to define and characterize early-life microbiota samples," Hall told GenomeWeb Daily News in an e-mail. The earliest parts of human life are a critical period in terms of the microbiome because at birth the human gut is completely bacteria-free, Hall explained, noting that the processes that follow birth and lead to microbial colonization are not fully understood. Having a better understanding of these processes could lead to treatments for diseases such as bacterial gastroenteritis, she said. This infectious disease is an increasing cause of infant death in the developing world, and the treatment involves antibiotics, but resistance to antibiotics is increasing and antibiotics also may reduce natural defenses against infection. Under this project, she will look to understand how antibiotics can disrupt these microbial communities, and search for probiotic bacteria.





{kind=link}
{kind=link}