- Oligonucleotides are Ligated by Chemical Reactions, Forming Novel Squaramide and Urea Linkages
- These Artificial Backbone Linkages in DNA Can Be Read-Through by Polymerases
- RNA Can Now Be Quantified by qPCR Without Enzymatic Ligation
Introduction
According to an expert review by Shuman, the discovery of DNA ligases in 1967 by the Gellert, Lehman, Richardson, and Hurwitz laboratories was a watershed event in molecular biology (reviewed by Lehman). DNA ligases play a key role in maintaining genome integrity, as they enzymatically join 3′-OH and 5′-PO4 termini to form a phosphodiester linkage. Essential for DNA replication and repair in all organisms, ligases were acknowledged in the 2015 Nobel Prize in Chemistry. These enzymes have become critical reagents in the development of DNA biotechnology, including cloning, gene profiling, molecular diagnostics, and sequencing methods. The numbers of annual publications on DNA ligation in PubMed for 1970—2017 shown here reflect their importance.
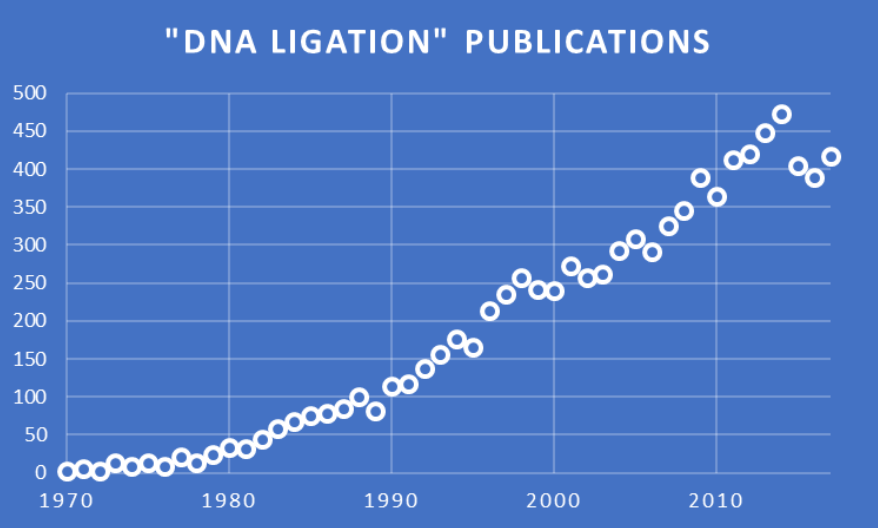 PubMed search and chart by Jerry Zon
PubMed search and chart by Jerry Zon
 Depiction of bacteriophage T4 infecting a bacterium
Depiction of bacteriophage T4 infecting a bacterium
DNA-templated DNA ligases can be used to distinguish single nucleotide polymorphisms (SNPs) among DNA sequences by exploiting the inefficient ligation of terminally mismatched oligonucleotides (oligos). This mechanism has been used for gene detection in the oligonucleotide ligation assay (OLA) and in the ligase chain reaction (LCR). Early studies of bacteriophage T4 DNA ligase showed that this enzyme could join DNA oligos hybridized to RNA templates, albeit with a substantially lower efficiency compared to DNA-templated reactions.
 Prof. Tom Brown, with permission
Prof. Tom Brown, with permission
Subsequently, RNA-templated ligation of DNA or RNA oligo probes has been used to generate molecules that can be amplified by PCR and the Qβ replicase, respectively. However, careful attention to NaCl and ATP concentrations is necessary, according to mechanistic studies by Nilsson et al. titled RNA-templated DNA ligation for transcript analysis. Susceptibility of enzymatic ligations to such factors has prompted researchers to explore the possibility of using purely chemical ligation as an alternative for ligase enzymes, i.e. non-enzymatic joining of oligonucleotides, for subsequent amplification by polymerases. Achieving this challenging goal has been recently exemplified by the innovative use of unnatural internucleotide linkages reported by Prof. Tom Brown and collaborators (Shivalingam et al.), as outlined in the following sections.
Backstory
According to Shivalingam et al., a wide variety of purely chemical methods for nucleic acid ligation can be used to obtain “artificial backbones” (i.e. non-phosphodiester internucleotide linkages) that are nevertheless recognized and read-through by DNA polymerases. These include amide, Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC), phosphoramidate (PA), phosphorothioate (PS), and thiol-thiol (disulfide-forming) reactions. However, they add, all of these face potential drawbacks, such as copper dependence (CuAAC), precursor handling (disulfide, PA, PS), slow ligation rates (PS), and poor read-through fidelity (disulfide).
Shivalingam et al. reasoned that these limitations warrant exploration of chemical ligation involving novel modified (mod) oligos and artificial backbones that synergize all of the advantageous properties: excellent fidelity, reversibility, and pre-activation. To this end, they describe a versatile ligation platform utilizing readily synthesized 3’- and 5’-amino mod oligos in conjunction with novel squaramide and urea internucleotide linkages, as outlined below.
 Word Cloud created by Jerry Zon
Word Cloud created by Jerry Zon
Design Strategy
The approach used by Shivalingam et al. begins with the synthesis of a pair of mod oligos, one with a 3’-amino group derived from the use of 3'-amino-dT CPG, and the other with a 5’-amino group derived from 5'-amino-dT-CE phosphoramidite, both of which are shown here and available from Glen Research.
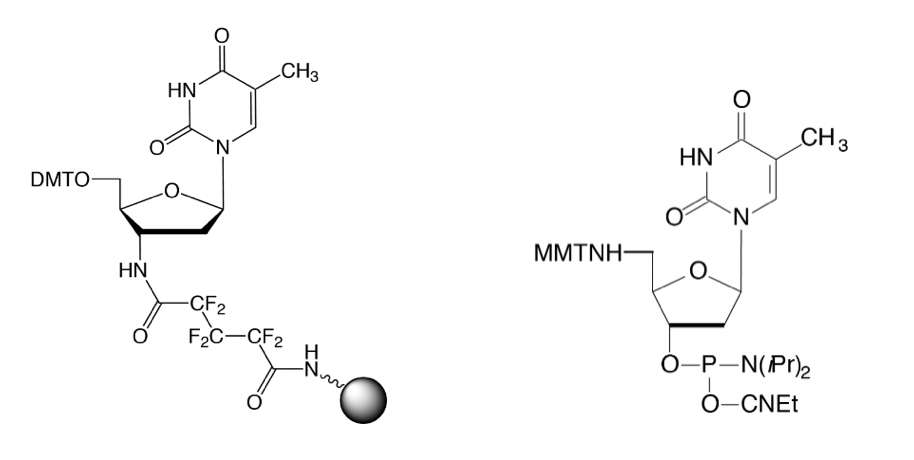 3'-Amino-dT CPG 5'-Amino-dT-CE phosphoramidite
3'-Amino-dT CPG 5'-Amino-dT-CE phosphoramidite
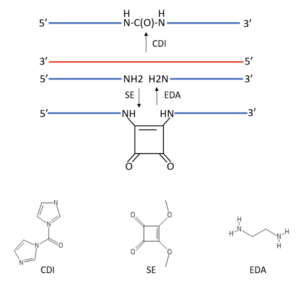 Scheme for the overall design strategy. Created by Jerry Zon
Scheme for the overall design strategy. Created by Jerry Zon
These mod oligos with terminal NH2 groups, which are blue in the overall design scheme shown below, are hybridized with a complementary DNA or RNA strand in red in the directional orientation shown. To generate artificial backbone linkages, two strategies were envisaged for chemical ligation. The first of these used 1,1’-carbonyldiimidazole (CDI) to form a urea linkage, based on earlier work of Kutterer & Just that demonstrated a urea linkage in a mod DNA oligo was well tolerated in hybridization with complementary DNA. The second strategy employed squarate ester (SE) to form a squaramide linkage, based on an earlier report by Sato et al. that a mod oligo with a squaramide linkage could hybridize with complementary DNA, although with diminished affinity.
Importantly, according to Shivalingam et al., the formation of urea or squaramide linkages by templated ligation have not been previously explored, and their read-through fidelity has not been either. Furthermore, the CDI and SE reagents used to generate these linkages are expected to become progressively less reactive upon each nucleophilic addition-elimination step, therefore potentially allowing each starting oligo to be pre-activated prior to ligation with another strand. The results briefly summarized in the next section include, among other things, the verification and utility of this two-step pre-activation scenario compared to a one-step, “one-pot” reaction.
Results and Discussion
Shivalingam et al. conducted extensive, systematic investigations of the design strategy outlined above. Readers interested in such details are referred to the journal publication and supplemental information. What follows is my synopsis of selected “technical takeaways” I believed were worth mentioning.
 One-Step Ligation: A 10-mer 3’-NH2 mod oligo and 30-mer 5’-NH2 mod oligo were hybridized to the complementary 30-mer DNA sequence s at pH 8.5. Under these conditions, the alkyl amine/ammonium ion equilibrium is shifted favorably to the amine form, while the templating strand significantly enhances the second nucleophilic addition-elimination step by reducing its concentration dependence.
One-Step Ligation: A 10-mer 3’-NH2 mod oligo and 30-mer 5’-NH2 mod oligo were hybridized to the complementary 30-mer DNA sequence s at pH 8.5. Under these conditions, the alkyl amine/ammonium ion equilibrium is shifted favorably to the amine form, while the templating strand significantly enhances the second nucleophilic addition-elimination step by reducing its concentration dependence.
Based on gel analysis, squaramide- and urea-linked oligonucleotide ligation proceeded well using SE and CDI (71% and 46% respectively). For the squaramide, ligation efficiency was dependent on pH (best to worst: 8.5 > 7.5 > unbuffered) and SE concentration (optimal = 5 mM). For the urea, CDI was used as a solid in large excess, making optimization of the urea ligation conditions challenging, as the imidazole byproduct buffers the system to neutral pH.
 Two-Step Ligation: Mass spectrometry (MS) of the above-mentioned one-step crude squaramide-formation reaction mixture showed that residual amino oligos were quantitatively converted to mono-squaramide monoesters. Therefore, pre-activation of either the 3'- or 5'-NH2 mod oligo was investigated before templated ligation to the second strand. Using identical conditions to the one-step ligation reaction and simple desalting with no further purification, pre-activation was straightforward.
Two-Step Ligation: Mass spectrometry (MS) of the above-mentioned one-step crude squaramide-formation reaction mixture showed that residual amino oligos were quantitatively converted to mono-squaramide monoesters. Therefore, pre-activation of either the 3'- or 5'-NH2 mod oligo was investigated before templated ligation to the second strand. Using identical conditions to the one-step ligation reaction and simple desalting with no further purification, pre-activation was straightforward.
By MS and gels analyses, no amino-oligo dimerization was observed and near-quantitative activation to mono-squaramide monoesters was achieved even at very high (100 μM) mod oligo concentrations. When added to the appropriate amino oligos, good conversion to the squaramide was observed within 15 min if a templating strand was present. The 5'-NH2 pre-activated oligo gave slightly lower ligation efficiency (80% vs. 88% for the pre-activated 3'-NH2 oligo), most likely due to the 3'-NH2 group being a more sterically hindered nucleophile.
 Polymerase Read-Through: Chemically ligated DNAs obtained from the CDI and SE reactions shown above were studied by qPCR using Phusion (exo+) and Taq (exo–) polymerases. The premise was that initial read-through of artificial backbones generating the unmodified complementary strand was rate-determining for the amount of generated PCR product. Detection of PCR product at lower cycles is therefore indicative of higher read-through efficiency.
Polymerase Read-Through: Chemically ligated DNAs obtained from the CDI and SE reactions shown above were studied by qPCR using Phusion (exo+) and Taq (exo–) polymerases. The premise was that initial read-through of artificial backbones generating the unmodified complementary strand was rate-determining for the amount of generated PCR product. Detection of PCR product at lower cycles is therefore indicative of higher read-through efficiency.
Both polymerases tolerated the urea linkage well, with PCR product fluorescence reaching a detectable level at cycles [“cycle threshold” (Ct)>
comparable to that of the native phosphodiester (control) linkage. Conversely, these polymerases were less tolerant of the squaramide linkage: △Ct control - squaramide = ~10 (Phusion) and ~14 (Taq) cycles, equivalent to ~1,000 and ~10,000-fold less PCR product respectively, assuming 100% PCR efficiency. Remarkably, Vent (exo–) gave comparable Ct values for control and the squaramide backbone.
 Fidelity of Polymerase Read-Through: After assessing the relative initial read-through efficiency of the above-mentioned polymerases, researchers then addressed the fidelity of dNTP incorporation into the product strand adjacent to the squaramide or urea linkages. PCR amplicons were generated using Vent (exo–) polymerase and synthetic squaramide- or urea-containing templates, cloned into a vector, and transformed in E. coli. Several colonies were randomly picked (n = 10 and 8 for squaramide and urea, respectively) and the recovered vectors were Sanger sequenced.
Fidelity of Polymerase Read-Through: After assessing the relative initial read-through efficiency of the above-mentioned polymerases, researchers then addressed the fidelity of dNTP incorporation into the product strand adjacent to the squaramide or urea linkages. PCR amplicons were generated using Vent (exo–) polymerase and synthetic squaramide- or urea-containing templates, cloned into a vector, and transformed in E. coli. Several colonies were randomly picked (n = 10 and 8 for squaramide and urea, respectively) and the recovered vectors were Sanger sequenced.
All colonies except one displayed correct base specificity adjacent to the artificial linkages. The one exception was a dT deletion directly adjacent to the site of the urea linkage, an infrequent occurrence (1/8). The urea linkage also displayed four other single-base mismatches, but these were non-conserved and occurred greater than 10 bases away from the modification site. For the squaramide, only one C-to-T mismatch mutation was observed 7 bases away from the modification.
Regeneration of 3’- and 5’-NH2 Mod Oligos: According to Sato et al., squaramide-containing oligos were susceptible to hydrolysis during ammonia-mediated nucleobase deprotection. This raised the possibility of excising the squaramide linkage and regenerating the 3’- and 5’-NH2 mod oligo starting materials under appropriate conditions. To evaluate this “chemical recycling” idea, a variety of nucleophiles [methylamine, ethanolamine, ethylenediamine (EDA), cysteamine and dithiothreitol>
were screened for their ability to cleave a squaramide linkage in 1 h at 55 °C, as depicted in the scheme for the overall design strategy shown above.
 Graphical abstract for recycling of 3’- and 5’-NH2 mod oligos by chemical reactions of a squaramide linkage in probes after non-enzymatic ligation.
Graphical abstract for recycling of 3’- and 5’-NH2 mod oligos by chemical reactions of a squaramide linkage in probes after non-enzymatic ligation.
Although thiols were ineffective, primary amines gave appreciable squaramide backbone cleavage. EDA was the most effective reagent. The identities of the postulated 3’- and 5’-squaramide-EDA oligo-EDA adducts were confirmed by UPLC-MS. Following the reaction over time indicated that the rate determining step to recover the starting material was removal of the squaramide-ethylenediamine adduct from the 3'-NH2 terminus of the oligo. Optimized cleavage conditions involved the addition of 50% aqueous EDA to an equal volume of aqueous oligo sample. Heating for 3 h at 55 °C gave ~95% cleavage of the linkage.
Confirmation that the starting material had been recovered was demonstrated by desalting the sample by gel-filtration (which also removed EDA), freeze-drying, and re-ligating the oligos using one-pot ligation conditions. The result was a comparable coupling yield to the one observed during the original ligation optimization.
Strategy for RNA Detection by qPCR: To demonstrate the utility of this enzyme-free ligation platform, Shivalingam et al. focused on RNA detection. Reverse-transcriptase-mediated PCR (RT-qPCR) is the current gold standard for RNA detection, but according to Shivalingam et al., the methodology has limitations. Namely, (1) the priming strategy used for initial RNA-to-cDNA conversion (e.g. random hexamers vs. poly(dT) primers) is known to influence the results obtained; (2) genomic DNA contamination remains an issue even with DNase-treated RNA samples; and (3) the identity of the PCR product needs to be validated during or post-PCR, typically using an oligo that hybridizes to the PCR product.
As depicted in the scheme shown here, the ligation-dependent qPCR design by Shivalingam et al. requires two mod DNA oligos (blue) to hybridize to RNA (red) next to each other in buffered, reagent-free conditions to form the template that will be used for qPCR. This validates the identity of the target RNA prior to qPCR directly from the unmodified source sample. More importantly, no polymerase-priming strategy is required, as the RNA-to-DNA conversion relies solely on hybridization. For these reasons, enzymatic bias in template read-through and/or secondary structure interference are minimized.
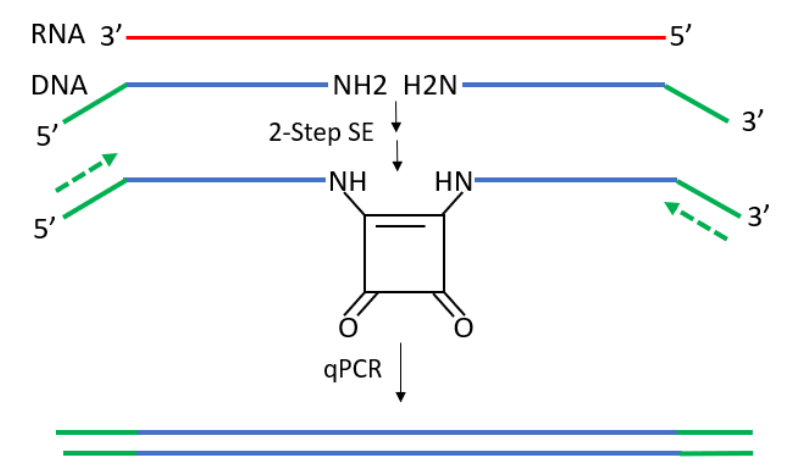 Scheme for detection of RNA by qPCR. Created by Jerry Zon
Scheme for detection of RNA by qPCR. Created by Jerry Zon
Since the DNA oligos will be directly used for qPCR, the introduction of sequence “tails” (solid-green) not found naturally in the genome enable the design of primers that are highly specific to the DNA template. In designing such a pair of RNA-detecting DNA oligos, each oligo was comprised of a 20-mer region that hybridized to the RNA target and a 10-mer 5'-tail for PCR primer (dashed green) binding. The two 20-mer regions were designed to minimize self-complementary, which would otherwise lead to template-independent ligation, and to be highly specific to the target as determined by BLAST alignment against a reference database. The 10-mer sequences were designed to be noncomplementary to the target and provide a relatively unstructured tail for PCR primers to efficiently bind to for amplification, as determined by mfold.
Proof-of-Concept for RNA Detection by qPCR: Shivalingam et al. initially designed DNA mod oligos that targeted a 100-mer RNA. The 3'-NH2 oligo was pre-activated with SE and kept as a stock solution, while the 5'-NH2 oligo DNA was left un-activated. The two mod oligos were then mixed with the RNA template (3 μM to 300 pM, five-orders of magnitude) and the ligation was allowed to proceed for 15 min before analysis of the product by qPCR. Specific RNA target amplification was observed, with non-specific primer-only (“primer dimer”) amplification limiting the sensitivity of the system.
In light of these promising results, researchers sought to push the limits of the system. Heat-activated CleanAmp® dNTPs (depicted here) were used to reduce unwanted primer-dimer amplification, as heat-activated Vent (exo–) polymerase was not commercially available. Furthermore, a more complex system comprising of a 2,664-mer target RNA prepared through in vitro transcription spiked into total RNA extracted from MCF-7 cells was also investigated.
 Taken from a TriLink CleanAmp® poster, which can be consulted for technical details.
Taken from a TriLink CleanAmp® poster, which can be consulted for technical details.
The target RNA was titrated into the total RNA, enabling for determination of the dynamic range and detection limits of the system (0.3 μM to 30 pM, five-orders of magnitude). Gratifyingly, the qPCR efficiency (110%, R2= 0.995) was within the generally accepted criteria of 95–110%, yielding comparable results to conventional RT-qPCR of the same samples. RNA detection was ultimately limited by ligation in the absence of the RNA target. Shivalingam et al. suggest that this could potentially be addressed by decreasing the mod oligo concentration in the ligation reaction; however, sufficient mod oligo must be present to ensure the equilibrium is favored for hybridization to low concentrations of RNA targets.
Conclusions
 Shivalingam et al. conclude that the squaramide and urea ligation platforms “expand the scope of polymerase-compatible nucleic acid assembly and crucially provide greater flexibility in this process than previously reported methods.” This flexibility includes one-step (one-pot) chemical ligation to generate either of two different linkages: (1) a urea linkage that multiple polymerases can readthrough with excellent speed, or (2) a squaramide linkage that offers accurate read-through under selective conditions and whose formation can be reversed. In this latter case, the linkage can also be formed from stable pre-activated intermediates in mild buffered conditions in the absence of small molecule reagents.
Shivalingam et al. conclude that the squaramide and urea ligation platforms “expand the scope of polymerase-compatible nucleic acid assembly and crucially provide greater flexibility in this process than previously reported methods.” This flexibility includes one-step (one-pot) chemical ligation to generate either of two different linkages: (1) a urea linkage that multiple polymerases can readthrough with excellent speed, or (2) a squaramide linkage that offers accurate read-through under selective conditions and whose formation can be reversed. In this latter case, the linkage can also be formed from stable pre-activated intermediates in mild buffered conditions in the absence of small molecule reagents.
 Shivalingam et al. add that “the diversity of this platform lends itself to many [cited>
Shivalingam et al. add that “the diversity of this platform lends itself to many [cited>
applications including DNA encoded-libraries, aptamer proximity-ligation and DNA nano-construct assembly.” Here, they have demonstrated its use in RNA detection, where it performs comparably to RT-qPCR, while offering advantages such as removing RNA polymerase bias, faster speed of execution, and greater inherent target specificity.
Personally, and speaking as an organic chemist working in the field of nucleic acids research, it is amazing to see how researchers such as Shivalingam et al. can push the boundaries of traditional nucleic acid chemistry and biochemistry by incorporating non-native structural moieties. One can only wonder what will be achieved by future chemical manipulations.
Your comments are welcomed, as usual.
Addendum
It’s worth noting that DNA ligation Searches in Google Scholar for the phrase “chemical ligation” with the terms “DNA” or “RNA” give more than 10,000 items, and thus indicate a large scientific field of interest for readers to peruse. A 2018 report by Abe & Kimura titled Chemical ligation reactions of oligonucleotides for biological and medicinal applications is one of many examples of recent literature involving non-native linkages in RNA. These researchers designed a new RNA interference (RNAi) syste, called intracellular buildup RNAi (IBR-RNAi), wherein RNA fragments are built up into active small-interference RNA (siRNA) in cells through a chemical ligation reaction. Using nucleophilic phosphorothioate (PS) and electrophilic iodoacetyl [ICH2C(O)>
moieties as reactive functional groups for the ligation, RNAi effects were achieved.
 Word cloud created by Jerry Zon
Word cloud created by Jerry Zon
Another highly cited item of historical note is the 1991 publication by Shabarova et al. titled Chemical ligation of DNA: the first non-enzymatic assembly of a biologically active gene. An artificial gene comprising of 183 base pairs was assembled by DNA template-directed condensation of 35- to 53-membered ODNs with cyanogen bromide (BrCN) as a condensing agent to generate native phosphodiester linkages. The reaction was complete within several minutes at 0°C in buffer, and the resulting mini-gene was cloned and expressed in vivo and in vitro.





