- 10 Million People Worldwide Have Parkinson’s Disease (PD)
- There is Currently No Cure for PD
- Treating PD with An Antisense Oligonucleotide is Now Clinically Feasible
 Parkinson’s disease (PD), a physically devastating neurodegenerative disorder of the central nervous system (CNS), is the second most common disease in the U.S. after Alzheimer’s. According to statistics provided by the Parkinson’s Foundation, more than 10 million people are living with PD worldwide in 2020, including nearly one million people in the U.S. Approximately 60,000 Americans are diagnosed with PD each year. The motor symptoms of the disease the result of cell death in the substantia nigra region of the midbrain, which leads to insufficient amounts of the neurotransmitter dopamine.
Parkinson’s disease (PD), a physically devastating neurodegenerative disorder of the central nervous system (CNS), is the second most common disease in the U.S. after Alzheimer’s. According to statistics provided by the Parkinson’s Foundation, more than 10 million people are living with PD worldwide in 2020, including nearly one million people in the U.S. Approximately 60,000 Americans are diagnosed with PD each year. The motor symptoms of the disease the result of cell death in the substantia nigra region of the midbrain, which leads to insufficient amounts of the neurotransmitter dopamine.
There is some palliative care for PD, but considerable research is now aimed at finding improved treatments or a potential cure. Nucleic acid-based strategies are prominent among the new approaches for better treatment of the disease, as discussed in previous Zone blogs. Recently, a News & Views article in the prestigious Nature stated that new findings reported by Qian et al. on June 24, 2020 hold “huge promise for our ability to use cell-conversion strategies to treat neurodegenerative diseases.” The report, titled Reversing a model of Parkinson’s disease with in situ converted nigral neurons, describes work by a large team of collaborators led by Xiang-Dong Fu, Professor of Cellular and Molecular Medicine at the University of California, San Diego, La Jolla, CA. A brief synopsis of this study is provided here, following a short introduction.
Introduction
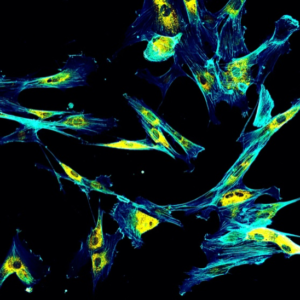 Immunofluorescence confocal imaging of fibroblasts with endoplasmic reticulum in yellow and cytoskeleton in cyan.
Immunofluorescence confocal imaging of fibroblasts with endoplasmic reticulum in yellow and cytoskeleton in cyan.
According to Qian et al., regenerative medicine holds great promise for the treatment of disorders marked by cell loss, adding that most in vivo reprogramming of cells relies on lineage-specific transcription factors. By contrast, the Fu team (Xue et al.) previously identified a new role for polypyrimidine tract binding protein (PTB), an RNA-binding protein capable of controlling neuronal induction and maturation. The Fu team demonstrated efficient conversion of fibroblasts to functional neurons by depletion of PTB, and went on to describe (Hu et al.) PTB and its structural homologs as “master regulators of neuronal fate in mammals.”
 Silver-stained microscopic image of a spinal cord neuron. The neurofibrils stained with silver nitrate can be seen in both the soma and dendrites of the neuron.
Silver-stained microscopic image of a spinal cord neuron. The neurofibrils stained with silver nitrate can be seen in both the soma and dendrites of the neuron.
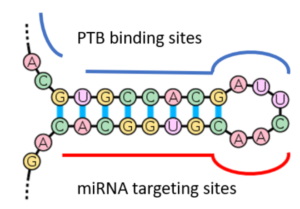 Depiction of a proposed (Xue et al.) model indicating PTB-mediated opening of a stem-loop in RNA that facilitates miRNA targeting. Drawn by Jerry Zon.
Depiction of a proposed (Xue et al.) model indicating PTB-mediated opening of a stem-loop in RNA that facilitates miRNA targeting. Drawn by Jerry Zon.
More specifically, Xue et al. and Hu et al. showed that PTBs have a function in the regulation of microRNA (miRNA) functions, suppressing or enhancing miRNA targeting by competitive binding on target mRNA or altering local RNA secondary structure, as depicted here. A key event during neuronal induction is the relief of PTB-mediated blockage of miRNA action, which de-represses a large array of neuronal genes, including miR-124—the most abundant miRNA in adult brain—and multiple neuronal-specific transcription factors in non-neuronal cells. This converts a negative feedback loop to a positive one that elicits cellular reprogramming to the neuronal lineage.
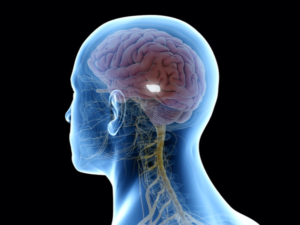 3D-rendered medically accurate illustration of the substantia nigra (white region).
3D-rendered medically accurate illustration of the substantia nigra (white region).
Based on these earlier findings, Qian et al. investigated a strategy to directly convert astrocytes to dopaminergic (DA) neurons in the substantia nigra region of midbrain (illustrated here) in which loss of dopaminergic neurons is associated with development of PD. As will be outlined in the following sections, Qian et al. used a chemically induced mouse model of PD to show that dopamine-producing neurons induced by PTB depletion potently restore striatal dopamine, reconstitute the nigrostriatal circuit, and reverse PD-like motor phenotypes. Given the emerging power of antisense oligonucleotides (ASOs) in modulating brain disorders, reviewed elsewhere, Qian et al. also provide evidence for the use of ASOs directed against PTBP1 (the gene that encodes PTB) as a feasible, single-step strategy for treating Parkinson’s disease and perhaps other neurodegenerative diseases.
Results
PTB-Regulated Signaling Loops in Astrocytes
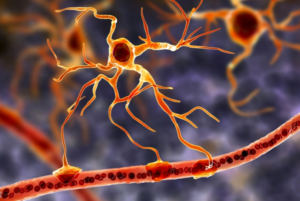 3D illustration of astrocytes, which connect neuronal cells to blood vessels.
3D illustration of astrocytes, which connect neuronal cells to blood vessels.
Glial cells are a class of cells that surround neurons, providing support for and insulation between neurons. Among the types of glial cells, astrocytes (illustrated here) offer several advantages for in vivo reprogramming in the brain. According to Qian et al., astrocytes are abundant, proliferate upon injury, and highly plastic with regards to cell fate.
As previously established in fibroblasts (Xue et al.), PTB suppresses a neuronal induction loop, in which miR-124 inhibits the RE1-silencing transcription (REST) repressor that suppresses many neuronal genes, including the gene encoding miR-124. Downregulation of PTB induces expression of its homolog, nPTB, which suppresses the transcription activator BRN2—a master regulator of neuronal differentiation—and miR-9, both of which are required for neuronal maturation. To investigate this cascade in the conversion of astrocytes to neurons, Qian et al. used mouse and human fetal astrocytes. RT-qPCR results confirmed that astrocytes can be converted to neurons by PTB knockdown alone in both mice and humans.

Efficient Astrocyte Conversion In Vitro
To demonstrate the functionality of astrocyte-derived neurons, Qian et al. transduced mouse astrocytes with a lentivirus expressing a small (aka short) hairpin RNA (shRNA) inhibitor against Ptbp1 (shPTB) via a RNA interference (RNAi) mechanism. The following results were obtained:
- After four weeks, 50 to 80% of shPTB-transduced cells showed neuronal morphology and stained positive for two neuronal markers (TUJ1 and MAP2), whereas transduction with control virus did not cause expression of these markers.
- RNA-sequencing (RNA-seq) analysis was performed before, during, and after the conversion of astrocytes to neurons. During conversion, typical astrocyte genes were suppressed, whereas neuronal genes were induced. Importantly, midbrain astrocytes gave rise to neurons expressing many dopaminergic (DA) neuron-specific genes.
 γ-Aminobutyric acid (GABA). Taken from commons.wikimedia.org and free to use.
γ-Aminobutyric acid (GABA). Taken from commons.wikimedia.org and free to use.Mouse and human astrocyte-derived neurons were positive for the neuronal lineage markers NeuN and NSE, and most of the expressed markers of glutamatergic or γ-aminobutyric acid (GABA)-containing neurons.
- Patch clamp recording six to eight weeks after conversion showed currents of voltage-gated sodium and potassium channels and repetitive action potential firing in neurons derived from both mouse and human astrocytes. No neuronal electrophysiological properties were detectable in astrocytes transduced with control virus.
 γ-Aminobutyric acid (GABA). Taken from commons.wikimedia.org and free to use.
γ-Aminobutyric acid (GABA). Taken from commons.wikimedia.org and free to use.
All these results, as well as additional findings (see Qian et al.), demonstrate a one-step conversion of astrocytes to functional neurons by RNAi-mediated depletion of PTB via treatment with lentivirus-encoded shRNA targeting PTB (shPBT). The next step was to test whether this reprograming could be achieved in vivo.
Generating New DA Neurons in Mouse Brain
 3D illustration of a molecular model of adenovirus, a DNA-virus.
3D illustration of a molecular model of adenovirus, a DNA-virus.
To directly reprogram astrocytes into neurons in mouse brain, Qian et al. designed an adeno-associated virus (AAV; illustrated here) vector to express shPTB (AAV-shPTB) and a corresponding empty AAV vector lacking shPTB (AAV-empty) as control. Lineage tracing was enabled by insertion of a red fluorescent protein (RFP) coding sequence 5’ to the shPTB hairpin that was activated in cells expressing Cre recombinase.
Focusing on the substantia nigra of the midbrain where DA neurons reside, it was found that RFP+ cells (of the type shown here) were almost completely absent 10 weeks after injecting either AAV-empty or AAV-shPTB in wild-type mice at 1 to 2 months of age, a developmental stage in which astrocytes have already lost their neurosphere-generating potential in the midbrain. By contrast, RFP was expressed in response to AAV-shPBT in mice expressing Cre recombinase. RFP was detected in ~1% of NeuN+ neurons, demonstrating minimal Cre expression in endogenous neurons in young adult mice. By contrast, 3 weeks after AAV-shPTB injection, ~20% of RFP+ cells expressed NeuN. The percentage of RFP+NeuN+ cells increased to ~60% by 5 weeks, and reached ~80% by 10 weeks.
 Confocal microscopy 3D projection of medium spiny neurons (MSNs) in the mouse striatum. The neurons were labeled using the matrisome MSN mouse Gpr101-Cre in combination with a dtTomato (red fluorescent protein, RFP) reporter. Taken from commons.wikimedia.org and free to use.
Confocal microscopy 3D projection of medium spiny neurons (MSNs) in the mouse striatum. The neurons were labeled using the matrisome MSN mouse Gpr101-Cre in combination with a dtTomato (red fluorescent protein, RFP) reporter. Taken from commons.wikimedia.org and free to use.
Qian et al. then monitored the gradual appearance of DA neurons among RFP-labelled cells from 3 to 12 weeks after AAV-shPTB injection in the midbrain. On the basis of staining with the DA neuron markers DOPA decarboxylase (DDC) and tyrosine hydroxylase (TH; see below), there was a progressive increase in the number of converted DA neurons, which reached 30–35% of RFP+ cells 12 weeks after injection. Progressive functional maturation of new DA neurons within the dopaminergic neuron-containing domain of the midbrain was also confirmed by patch clamp measurements.
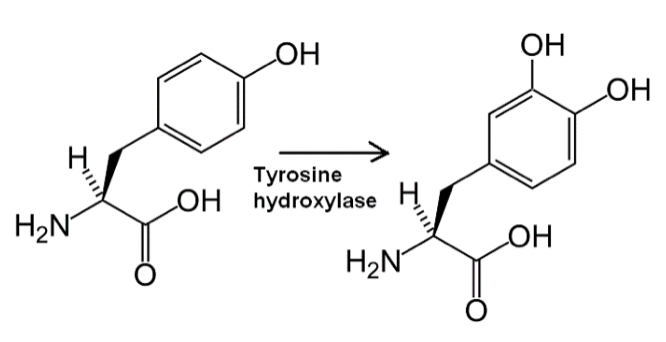 Tyrosine hydroxylase is the enzyme responsible for catalyzing the conversion of the amino acid L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA) using molecular oxygen (O2), as well as iron (Fe2+) and tetrahydrobiopterin as cofactors. L-DOPA is a precursor for dopamine, which is in turn a precursor for the important neurotransmitters norepinephrine (noradrenaline) and epinephrine (adrenaline). Taken from commons.wikimedia.org and free to use.
Tyrosine hydroxylase is the enzyme responsible for catalyzing the conversion of the amino acid L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA) using molecular oxygen (O2), as well as iron (Fe2+) and tetrahydrobiopterin as cofactors. L-DOPA is a precursor for dopamine, which is in turn a precursor for the important neurotransmitters norepinephrine (noradrenaline) and epinephrine (adrenaline). Taken from commons.wikimedia.org and free to use.
Replenishing Lost DA Neurons in a Disease Model
 6-Hydroxydopamine (6-OHDA). Taken from wikipedia.org and free to use.
6-Hydroxydopamine (6-OHDA). Taken from wikipedia.org and free to use.
Following the successful generation of DA neurons, Qian et al. investigated whether these neurons could reconstitute an injured nigrostriatal pathway. They selected a widely used model of PD in mouse in which DA neurons are efficiently ablated by 6-hydroxydopamine (6-OHDA, shown here), a dopamine analogue toxic to DA neurons. Although this model does not recapitulate all essential features of PD pathogenesis, according to Qian et al., it does result in a critical endpoint—the loss of neurons in the substantia nigra and depletion of striatal dopamine.
One month after 6-OHDA injection into one side of the medial forebrain bundle, the researchers observed unilateral loss of TH+ cell bodies in the midbrain, accompanied by a marked increase in glial fibrillary acidic protein (GFAP)-producing (GFAP+) astrocytes, indicative of the expected astrocytic response. One month after the lesion, they injected AAV-empty or AAV-shPTB in the lesioned side and observed increased RFP+TH+ cell bodies around 10–12 weeks later with AAV-shPTB, but not with AAV-empty.
Restoration of Striatal Dopamine
 Qian et al. then investigated whether AAV-shPTB-induced neurons would restore dopamine levels in the striatum by preparing extracts and quantifying dopamine levels using high-performance liquid chromatography (HPLC). Samples were spiked with known quantities of dopamine to define the elution position and establish the relationship between signal intensities and the amount of dopamine. They detected similar amounts of dopamine in both sides of uninjured mice and found that 6-OHDA lesion reduced dopamine to about 25% of the normal level. Treatment with AAV-shPTB, but not with AAV-empty, markedly increased the dopamine level compared with lesioned striatum, reaching ~65% of the uninjured level.
Qian et al. then investigated whether AAV-shPTB-induced neurons would restore dopamine levels in the striatum by preparing extracts and quantifying dopamine levels using high-performance liquid chromatography (HPLC). Samples were spiked with known quantities of dopamine to define the elution position and establish the relationship between signal intensities and the amount of dopamine. They detected similar amounts of dopamine in both sides of uninjured mice and found that 6-OHDA lesion reduced dopamine to about 25% of the normal level. Treatment with AAV-shPTB, but not with AAV-empty, markedly increased the dopamine level compared with lesioned striatum, reaching ~65% of the uninjured level.
Reversing PD-Relevant Motor Phenotypes
 The ability of AAV-shPTB to restore motor function to mice with 6-OHDA lesions was tested with three common behavioral tests, two based on drug-induced rotation, and one based on spontaneous motor activities. Contralateral rotation induced by apomorphine and ipsilateral rotation triggered by amphetamine were markedly increased following lesion with 6-OHDA. Both phenotypes were progressively restored to nearly wild-type levels within three months after AAV-shPTB treatment. No correction was recorded in mice treated with AAV-empty or with non-specific AAV-shGFP.
The ability of AAV-shPTB to restore motor function to mice with 6-OHDA lesions was tested with three common behavioral tests, two based on drug-induced rotation, and one based on spontaneous motor activities. Contralateral rotation induced by apomorphine and ipsilateral rotation triggered by amphetamine were markedly increased following lesion with 6-OHDA. Both phenotypes were progressively restored to nearly wild-type levels within three months after AAV-shPTB treatment. No correction was recorded in mice treated with AAV-empty or with non-specific AAV-shGFP.
As PD and most other types of neurodegenerative diseases show age-dependent onset, Qian et al. extended the study from relatively young (two-month-old) mice to one-year-old mice, an age comparable to the age of onset of PD in humans. While the behavioral benefits of AAV-shPTB transduction following apomorphine-induced rotation did not reach statistical significance—perhaps owing to relatively unstable phenotype scored by this assay in aged animals—a substantial improvement was recorded with the limb-use asymmetry test. It was concluded that “these observations point to an age-related decrease in neuronal reprogramming, a critical challenge to be met in future studies.”
ASO-Based Neuronal Conversion and Rescue
According to Qian et al., the PTB-regulatory loop is self-reinforcing once it is triggered by initial PTB knockdown. In response to a reduction in PTB, miR-124 becomes more efficient at targeting REST (owing to the ability of PTB to directly compete with the miRNA-targeting site in the 3′-untranslated region of the mRNA encoded by REST), resulting in decreased levels of REST, which drives further de-repression of miR-124 and thus further suppression of PTB. Importantly, this suggests that transient suppression of PTB might be sufficient to generate new neurons through ASO-mediated PTB mRNA degradation by intranuclear RNase H.
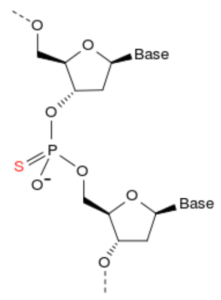 Phosphorothioate linkage in DNA. Taken from commons.wikimedia.org and free to use.
Phosphorothioate linkage in DNA. Taken from commons.wikimedia.org and free to use.
To test this possibility, Qian et al. synthesized and screened PTB ASOs containing a phosphorothioate backbone (depicted here) and a 3′ fluorescein. An ASO targeting GFP served as control. Three PTB ASOs, but not GFP ASO, reduced PTB expression upon transfection into mouse astrocytes. The best targeting PTB ASO (5′-GGGTGAAGATCCTGTTCAATA-3′), but not GFP ASO, induced expression of multiple neuronal markers (TUJ1, MAP2, NSE, and NeuN) after 5 weeks. A fraction of converted neurons was dopaminergic (DA), as indicated by TH staining. Patch clamp recording showed that these in vitro-converted neurons were functional.
Next, PTB ASO or control GFP ASO were injected into the midbrain of transgenic mice carrying a tamoxifen-inducible Cre. This was selectively expressed in astrocytes by first inducing Cre in the mice and then stereotactically injecting ASOs unilaterally into their substantia nigra 2 weeks later. PTB ASO converted a fraction of cells to NeuN+ neurons after 8 weeks, and to TH+ neurons after 12 weeks. Patch clamp recording demonstrated that these in vivo-converted neurons displayed functional neurophysiological properties. Most notably, PTB ASO, but not control GFP ASO, rescued the 6-OHDA lesion-induced phenotype 3 months after injection, on the basis of both apomorphine-induced rotation and ipsilateral touch bias tests.
Conclusions
In summary, Qian et al. demonstrated a one-step ASO-mediated strategy to convert brain astrocytes to functional neurons. The approach utilizes the genetic underpinnings of a neuronal differentiation program that is present but latent in astrocytes. By exploiting the regional specificity of neuronal reprogramming, midbrain astrocytes were efficiently converted into functional DA neurons that integrated into the nigrostriatal dopamine pathway.
By applying this approach to a chemically induced model of PD, the researchers were able to demonstrate partial replenishment of lost DA neurons and the restoration of striatal dopamine, which lead to reversal of motor deficits. Notably, these ASO-based experiments illustrate a potentially clinically feasible approach for treatment of patients with PD.
Qian et al. cautioned that “eventual application of this approach to humans will need to overcome many obstacles, including age-related limits of reprogramming, understanding potential adverse effects caused by local astrocyte depletion..., specifically targeting regions that harbor vulnerable neurons, and detecting potential side effects due to mistargeted neurons.” However, they added, “each of these objectives can now be addressed experimentally to develop this promising therapeutic strategy—one that may be applicable to not only PD, but also other neurodegenerative disorders.”
 The Zone is impressed by these promising findings, which provide even more indications for the utility of nucleic acid-based therapeutics. It is time to find a cure for PD.
The Zone is impressed by these promising findings, which provide even more indications for the utility of nucleic acid-based therapeutics. It is time to find a cure for PD.
Your comments are welcomed, as usual.
Addendum
 After this blog was written, there was a July 1, 2020, publication by Elitt et al. in Nature that reported remarkable findings in a mouse model on the single-dose ASO treatment of Pelizaeus-Merzbacher disease (PMD), a disease characterized by extensive loss of myelinating oligodendrocytes (illustrated here) in the CNS. Symptoms typically present in early infancy or childhood and include spasticity and cognitive dysfunction, with mortality often prior to adulthood. Preclinical efforts to extend lifespan have demonstrated only limited success, and no therapy has shown efficacy in patients.
After this blog was written, there was a July 1, 2020, publication by Elitt et al. in Nature that reported remarkable findings in a mouse model on the single-dose ASO treatment of Pelizaeus-Merzbacher disease (PMD), a disease characterized by extensive loss of myelinating oligodendrocytes (illustrated here) in the CNS. Symptoms typically present in early infancy or childhood and include spasticity and cognitive dysfunction, with mortality often prior to adulthood. Preclinical efforts to extend lifespan have demonstrated only limited success, and no therapy has shown efficacy in patients.
A single intracerebroventricular injection of an ASO in postnatal mice targeting proteolipid protein 1 (PLP1) fully restored oligodendrocyte numbers, increased myelination, improved motor performance, normalized respiratory function, and extended lifespan through an 8-month endpoint. Elitt et al. concluded that “[t>
hese results support the development of PLP1-suppression as a treatment for PMD.”





