- Exciting potential of direct sequencing of modified DNA
- Small holes with big promise but bigger challenges
- Paleogenomics: sequencing ancient DNA—how old can you go?
Sometimes small scientific meetings have big impacts on one’s impressions, which was certainly my experience at the 3rd Next-Generation Sequencing (NGS) conference in San Francisco on June 19-21, 2013. Of the many interesting presentations (click here for all speakers and abstracts), three completely different topics struck me the most: Pacific Biosystems’ uniquely powerful single-molecule real-time (SMRT) sequencing of modified DNA, Sequencing-pioneer Prof. David Deamer’s update on Nanopore’s advances and challenges, and the new field of Paleogenomics involving sequencing old DNA. With apologies to all of the other speakers, and admitting personally biased selection, here are my comments about these three topics.
Pacific Biosystems: direct sequencing of modified DNA
 Dr. Jonas Korlach co-invented SMRT technology with Stephen Turner, Ph.D., PacBio Founder and Chief Technology Officer, when the two were graduate students at Cornell University. Dr. Korlach joined PacBio as the company's eighth employee in 2004. Dr. Korlach was appointed Chief Scientific Officer at PacBio in July, 2012.
Dr. Jonas Korlach co-invented SMRT technology with Stephen Turner, Ph.D., PacBio Founder and Chief Technology Officer, when the two were graduate students at Cornell University. Dr. Korlach joined PacBio as the company's eighth employee in 2004. Dr. Korlach was appointed Chief Scientific Officer at PacBio in July, 2012.
Pacific Biosystems (PacBio) deserves a lot of credit for being able to overcome numerous technical challenges facing commercialization of its SMRT sequencing system, which offers some uniquely powerful capabilities. (I’ll save a bit of time and space by refraining from describing how this complex system works, but I encourage you to take advantage of various videos and other technical information available at PacBio’s website.) In addition to providing amazingly long read lengths (up to 20kb) to facilitate genome assembly, SMRT sequencing gives data related to kinetics of nucleotide incorporation. Algorithms for differentiating rate of incorporation of A, G, C or T opposite a cognate nucleotide position in the template strand for various sequence contexts within the “footprint” of a DNA polymerase can also differentiate modified template positions. In other words, the average rate of incorporation of G opposite C is different than that opposite 5-methylcytosine (5-mC). This difference in kinetics allows direct determination of epigenetic methylation patterns in DNA, which was the focus of an excellent presentation by PacBio CSO Jonas Korlach. Direct epigenetic sequencing of 5-mC is completely novel and offers a significant advantage by obviating the need to carrying out so-called ‘bisulfite conversion chemistry’ prior to sequencing. Commercial kits are available for bisulfite conversion but require extra time, can be very tricky, and utilize more sample than may be available—especially for limited amounts of clinical biopsies.
I subsequently checked PacBio’s website and found a white paper pdf stating that unique kinetic characteristics have been observed for over 25 types of base modifications, such as those shown below and for these reasons:
- Human epigenetic markers in addition to 5-mC include 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC).
- Bacterial epigenetic markers for regulating DNA replication and repair and transcription regulation include 6-methyladenosine (6-mA), while bacterial identity markers for affecting host-pathogen interactions include 6-mA, 4-methylcytosine (4-mC), and 5-mC.
- Products of DNA damage include 8-oxoguanine (8-oxoG) and 8-oxoadenine (8-oxoA).
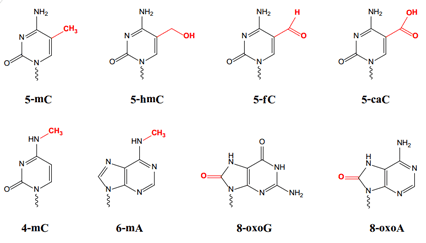 Molecular structures and abbreviations for modified bases directly identifiable by SMRT sequencing (taken from PacBio white paper).
Molecular structures and abbreviations for modified bases directly identifiable by SMRT sequencing (taken from PacBio white paper).
Especially exciting to me was Dr. Korlach’s brief mention at the end of his talk that SMRT could be used for direct sequencing of phosphorothioate (PS) linkages in DNA. While “man-made” PS modifications in synthetic DNA are well known, naturally occurring PS-DNA is a relatively recent—and quite surprising—discovery still being elucidated. A 2013 review (click here for pdf) of this novel and fascinating type of naturally occurring modified DNA states that “physiological PS modification is widespread in bacteria and occurs in diverse sequence contexts and frequencies [approximately 300 – 3,000 PS per 106 nucleotides>
in different bacterial genomes, implying a significant impact on bacteria.” Bacterial PS-DNA has been shown to be introduced by a post-replicative biochemical pathway associated with a cluster of five genes, and is implicated in site-specific restriction and, more recently, chemical reducing capacity to protect bacteria against peroxide. PS linkages in DNA can have SP or RP stereochemistry at the phosphorus as shown below; however, all bacterial PS-DNA examined to date occurs in the RP form.
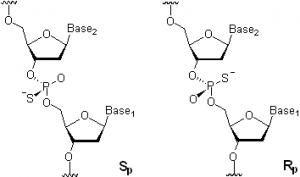 Generalized molecular structure of SP and RP PS-DNA linkages (taken from RS Phosphorothioates Wikipedia).
Generalized molecular structure of SP and RP PS-DNA linkages (taken from RS Phosphorothioates Wikipedia).
I later contacted Dr. Korlach to get more information about PS sequencing by SMRT and he referred me this video (~17 minutes) and conference abstract. In response to my question about whether SMRT sequencing could differentiate SP from RP stereochemistry, he replied that he and his collaborators have looked at this possibility but he couldn’t comment at this time because the work was ongoing and would be published in the future.
While awaiting publication of those findings, it’s interesting—I think—to speculate about other applications of SMRT direct sequencing of modified DNA. One intriguing possibility is determining the extent of, and genomic loci for, 5-fluoro-2'-deoxyuridine incorporation into DNA that heretofore has only been studied using indirect methods to decipher mechanisms of action of various 5-fluoropyrimidine anticancer agents.
What other possible applications of SMRT direct sequencing of modified DNA can you suggest? (Please include in the comments section.)
Nanopore sequencing: small holes with big promise but bigger challenges
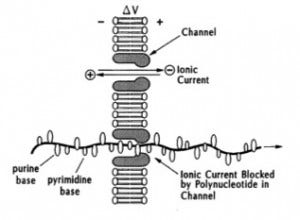
When I presented a Church, Deamer, Branton et al. patent that broadly describes nanopore sequencing of DNA (see below) to my former marketing colleagues at Applied Biosystems Inc. (ABI) in 1998, they enthusiastically asked “how soon can we sell a nanopore sequencer?” After I told them the patent was prophetic and had no actual data, they disappointedly said “too bad, let us know when it’s ready.” Well, it’s now 15 years later, and many folks like me are still waiting for that commercialization date, despite hundreds of publications on many different variations of the basic concept.
Consequently, I attended Prof. David Deamer’s presentation with the hope of learning when some type of nanopore sequencer would finally be introduced by any one of several companies in this space—notably Oxford Nanopore Technologies (ONT), whose stellar Technology Advisory Board includes Prof. Deamer.
 David W. Deamer is a Research Professor in the Department Chemistry & Biochemistry at UC Santa Cruz where his primary research area concerns the manner in which linear macromolecules traverse nanoscopic channels.
David W. Deamer is a Research Professor in the Department Chemistry & Biochemistry at UC Santa Cruz where his primary research area concerns the manner in which linear macromolecules traverse nanoscopic channels.
Prof. Deamer presented an excellent update starting with a stylized version of his original lab notebook sketch of the technology (see below). He then discusses some of the incremental progress—and many remaining challenges—for nanopore sequencing (check out reviews by Dunbar et al and others by searching “nanopore sequencing” on PubMed). He concluded with a description of recent results obtained by a group led by Prof. Mark Akeson, his long-time colleague and collaborator at UCSC. Among various innovations, a processive DNA polymerase is used to control translocation by ratcheting. Although the sequencing results presented were limited to only ~10 bases in a model oligonucleotide, a well-known and rather critical attendee—who I’ll keep anonymous—said during Q&A that “these were the most promising data I’ve seen so far.” That attendee then asked about ONT’s timeline for commercialization, to which Prof. Deamer said “he doesn’t speak for the company, but thinks that something might be introduced in another 6 months or so.”
Deptiction of nanopore sequencing method described in Church, Deamer, Branton et al. patent US 5,795,782.
At the risk of sounding like a pessimist, but based on my past experience where timelines for developing complex automated systems always took much longer than desired, I’d be very surprised if that “something” is launched by the end of 2013. Hopefully I am wrong so I’ll be on the look out just in case.
In the meantime, while we all await such an event, you can read about several thought-provoking nanopore sequencing-related topics:
- “Corporate spat” between ONT and Illumina in the nanopore sequencing space.
- Four lessons from IBM and Roche's failed nanopore sequencing collaboration.
- Base4 and Hitachi High-Tech recent announcement to develop a new nanopore sequencing system.
- Will “molecular hugging” graphene nanopores be the solution?
- Genia’s NanoTag sequencing approach.
- NabSys’ solid-state nanodetectors for probe-based “positional sequencing.”
Paleogenomics: sequencing ancient DNA—how old can you go?
Relative to evolutionary time-spans, the study of paleogenetics is not old—going back to 1963 and Linus Pauling; however, very, very old (aka “ancient”) DNA is now “sequenceable” using modern NGS technologies. Just how old is “ancient” and what is the projected age-limit for sequenceable DNA were two questions I had in mind at the outset of the presentation by Prof. Eske Willerslev, who has been a pioneer in this field.
 Prof. Eske Willerslev is a Danish evolutionary biologist at Copenhagen University and leader of the Ancient DNA and Evolution Group. He has received the Genius Award (Geniusprisen) of Danish Science journalists for his combination of groundbreaking research with an aggressive media strategy. Before becoming a scientist he lived for several years as a trapper in Siberia with his twin brother, anthropologist Rane Willerslev.
Prof. Eske Willerslev is a Danish evolutionary biologist at Copenhagen University and leader of the Ancient DNA and Evolution Group. He has received the Genius Award (Geniusprisen) of Danish Science journalists for his combination of groundbreaking research with an aggressive media strategy. Before becoming a scientist he lived for several years as a trapper in Siberia with his twin brother, anthropologist Rane Willerslev.
The presentation by Prof. Willerslev was rapidly delivered and jam-packed with snippets of results from numerous studies, which is another way of saying here that it was impossible for me to take notes from which to reconstruct a synopsis ex post facto using cited publications. On the other hand, I did get the following answers to my two probing questions.
Just how old is ‘ancient’ DNA?
Prof. Willersley said that a draft genome from a ~700 thousand years before present (~700k yr BP) horse bone found at Thistle Creek, Canada represents the oldest full genome sequence determined so far, and by almost an order of magnitude. This stunning—to me—achievement, which was published in Nature online several days after the conference and has received considerable attention because of the significance of its findings with regard to “recalibrating Equus evolution.” As stated in the abstract of this publication, “[f>
or comparison, we sequenced the genome of a Late Pleistocene horse (43 kyr BP), and modern genomes of five domestic horse breeds (Equus ferus caballus), a Przewalski’s horse (E. f. przewalskii) [pictured below>
and a donkey (E. asinus). Our analyses suggest that the Equus lineage giving rise to all contemporary horses, zebras and donkeys originated 4.0–4.5 million years before present (Myr BP), twice the conventionally accepted time to the most recent common ancestor of the genus Equus.”
 Przewalski's horse at Khustain Nuruu National Park in Mongolia. These horses are smallish and stocky in comparison to domesticated horses, with shorter legs that are often faintly striped, typical of primitive markings. The Przewalski's horse has 66 chromosomes, compared to 64 in all other horse species. All Przewalski horses in the world are descended from 9 of the 31 horses in captivity in 1945. These 9 horses were mostly descended from ~15 captured around 1900. The total number of these horses by the early 1990s was over 1,500.
Przewalski's horse at Khustain Nuruu National Park in Mongolia. These horses are smallish and stocky in comparison to domesticated horses, with shorter legs that are often faintly striped, typical of primitive markings. The Przewalski's horse has 66 chromosomes, compared to 64 in all other horse species. All Przewalski horses in the world are descended from 9 of the 31 horses in captivity in 1945. These 9 horses were mostly descended from ~15 captured around 1900. The total number of these horses by the early 1990s was over 1,500.
Interestingly, the aforementioned Nature publication reports using a combination of Illumina and Helicos sequencing, with the latter’s single-molecule sequencing capabilities providing an “advantageous complement” to the former’s data, as previously described. Since Helicos is now defunct, it will be interesting to see if such methodological complementarity can instead be provided by PacBio’s single-molecule sequencing.
How old can you go?
As for “how old can you go” and still get sequenceable DNA, Prof. Willerslev said at the conference that “1 or 2 million years old should be possible.” A subsequent article by Millar & Lambert in Nature News & Views entitled Towards a million-year-old genome confirmed this and noted—as expected—that degradation of DNA into ever shorter fragments begins rapidly after death by action of the body’s own enzymes, and then by action of enzymes from microorganisms. The overall rate of decay of DNA is also influenced by environmental conditions are such as pH and, of course, temperature, as shown in the following graph, which was entitled ‘survival of the coldest.’
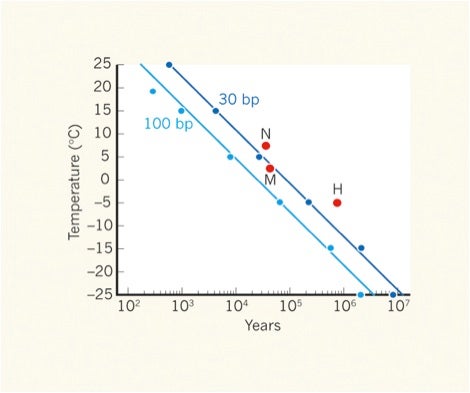 Plot of the rate of DNA decay vs. temperature for estimated half-lives of 30- and 100-base-pair (bp) DNA fragments. The estimated ages and temperatures of material used to recover the genomes of a Neanderthal (N), a woolly mammoth (M) and the horse fossil discovered at Thistle Creek, Canada (H) are shown [C. D. Millar & D. M. Lambert, Nature, Vol. 499, pp. 34-35 (2013)>
Plot of the rate of DNA decay vs. temperature for estimated half-lives of 30- and 100-base-pair (bp) DNA fragments. The estimated ages and temperatures of material used to recover the genomes of a Neanderthal (N), a woolly mammoth (M) and the horse fossil discovered at Thistle Creek, Canada (H) are shown [C. D. Millar & D. M. Lambert, Nature, Vol. 499, pp. 34-35 (2013)>
.
As always, your comments are welcomed.





{kind=link}