Previous blogs in the Zone have commented on the successful development of mRNA vaccines to blunt the spread of COVID-19 and, by extension, various other infectious diseases. Here, the Zone expands the discussion of mRNA vaccine applications to include cancer immunotherapy. We’ll look at four examples of general strategies directed at treating this large group of diseases that collectively accounted for 10 million deaths in 2020 alone:
- Shared Tumor Antigens
- Neoantigens
- Immunostimulants
- Chimeric Antigen Receptor T Cells
Before doing so, we’ll briefly mention some essential immunotherapy concepts for designing mRNA vaccines against cancer (Beck et al., 2021).
- First is the distinction between protect vs. attack: mRNA vaccines for COVID-19 and other infectious diseases protect people from viruses, i.e., prophylactic. In contrast, a cancer vaccine is a treatment given to patients with the hope that their immune system will be activated to attack tumor cells, i.e., they are primarily therapeutic.
- Second are the mechanisms by which tumor cells are attacked during immunotherapy: principally, through the induction of tumor-specific cytotoxic T-cells, by providing cancer antigens to antigen-presenting cells, and via the induction of tumor-specific B-cell responses (Beck et al. 2021).
- Third are the types of mRNA-delivered cancer antigens: either tumor-associated self-antigens (TAAs), such as differentiation antigens and overexpressed antigens, or truly tumor-specific antigens (TSAs) that are not subject to immune tolerance, such as neoantigens that form on cancer cells when certain mutations occur in tumor DNA.
These points will be touched upon in the following general strategic approaches.
1. mRNA Encoded Shared Tumor Antigen Cocktails
Because TAAs are non-mutated self-antigens, T-cell tolerance may contribute to the largely weak, clinically ineffective T-cell responses observed in previous vaccine trials (Melero et al., 2014).
This issue has been recently overcome by the development of a vaccine “cocktail” comprised of a fixed combination of shared TAAs. Termed “FixVac,” this strategy was reported by a large team of researchers for a Phase 1 clinical study of vaccinating patients with advanced stages of melanoma (Figure 1) (Sahin et al. 2020).
FIGURE 1. Picture of a skin melanoma. Approximately 2.5 cm (1 in) by 1.5 cm (0.6 in) Taken from commons.wikimedia.org and free to use. Credit: National Cancer Institute.
Briefly, four different in vitro transcribed mRNAs were individually prepared for separate liposomal formulation to deliver various doses of the following mixture of TAAs: (i.) New York esophageal squamous cell carcinoma 1, (ii.) melanoma-associated antigen A3, (iii.) tyrosinase, and (iv.) transmembrane phosphatase with tensin homology. These antigens all show restricted expression in normal tissue and high prevalence in, and immunogenicity against, melanoma. Patients expressing at least one of these four TAAs underwent eight vaccinations in a ‘prime/repeat boost’ protocol followed by optional continued monthly treatment.
An exploratory interim analysis showed that this melanoma FixVac (aka BNT111), alone or in combination with approved antibodies against the checkpoint-inhibitor protein PD1, mediates durable objective responses in patients with inoperable melanoma who had previously received checkpoint-inhibitor treatment. Clinical responses were accompanied by the induction of strong CD4+ and CD8+ T-cell immunity against the vaccine antigens.
Based on these promising findings, BioNTech announced in November 2021 that BNT111 received Fast Track Designation in advanced melanoma by the FDA.
2. mRNA Neoantigen Vaccines
As expertly reviewed elsewhere (Lang et al. 2022), mutated gene products can act as tumor neoantigens when their peptide products are broken down and presented as neoepitopes for recognition by CD4+ or CD8+ T cells. The number of somatic mutations in a tumor correlates with T-cell infiltration and is predictive of overall response rate and prolonged survival by immunotherapies across various cancer types.
Each cancer patient has a unique mutation profile and presents a unique composition of neoepitope–MHC complexes on their cancer cells. This is both an opportunity, i.e., highly specific targeted therapy, and a challenge, i.e., developing a highly personalized treatment for each patient. Achieving such treatment begins by obtaining a pair of genetic profiles from two individuals: one from a tumor biopsy, the other from a vial of healthy blood cells. Algorithms compare the DNA sequences of the two samples and produce a list of targets, each encoding a different mutant protein expressed by the cancer that is predicted to be useful in training the immune system to attack the disease (Lang et al. 2022).
BioNTech’s technical approach to individualized neoantigen-specific immunotherapy (iNeST) is said to be tailored to a specific patient’s tumor by using unmodified, pharmacologically optimized mRNA encoding up to 20 patient-specific neoantigens. The mRNAs are encapsulated in a proprietary intravenous mRNA-lipoplex delivery formulation designed to enhance stability and enable targeted delivery to dendritic antigen-presenting cells. Each cancer vaccine encodes for neoantigen candidates with the highest likelihood to help the immune system recognize cancer. For this purpose, it was necessary to develop a first-of-its-kind “on-demand” GMP manufacturing process.
Recently, this neoantigen mRNA vaccine strategy entered the clinic. In October 2021, BioNTech announced that it dosed the first patient in a Phase 2 trial of patients with high-risk colorectal cancer that are positive for circulating tumor DNA (ctDNA) after adjuvant treatment (NCT04486378).
Melanoma is also being clinically investigated by Moderna using its mRNA-personalized cancer vaccine (mRNA-4157, NCT 0389788) that involves neoantigens in a strategy very similar to this example employed by BioNTech.
3. Local mRNA Delivery of Immunostimulants
Systemic administration of recombinant cytokines is a clinically validated therapeutic strategy for cancer treatment. For example, IFN-α can directly induce tumor cell apoptosis (Chawla-Sakar et al. 2001), mediate anti-angiogenic effects on the tumor vasculature (Spaapen et al. 2014), leading to activation and maturation of dendritic cells, and promote the differentiation of effector T cells (Hervas-Stubbs et al. 2011). Although administration of two cytokines, IL-2 and IFN-α, are treatments for renal cell carcinoma and melanoma, respectively, their short half-lives require frequent administration, and systemic toxicities must be overcome by local administration (Zhang et al. 2018).
Interestingly, in preclinical tumor models, local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication in both treated tumors and distant or disseminated tumors, limiting the challenges of short half-lives and toxicity (Holtz et al. 2021). Briefly, IVT was used to prepare four different N1-methylpsedouridine-modified mRNAs, each encoding one of four murine cytokines: IL-12 single chain, interferon-α (IFN-α), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-15. Intratumor administration of a saline-formulated mixture of these four mRNAs led to effective antitumor activity involving multiple immune cell populations.
Several cancer cell lines were implanted in mice to obtain either a local tumor in the right and left flank or a disseminated tumor in the lungs. Remarkably, local delivery of the mRNA cytokine mixture into the left flank tumor led to its shrinkage and the reduction of tumors in the right flank or lungs. Combining the mRNAs with immunomodulatory antibodies enhanced antitumor responses, thus improving survival and tumor regression.
Last, the corresponding human-specific mRNA-encoded cytokines were expressed in vivo and induced in downstream pharmacodynamic responses from analyses of blood cell samples from patients with advanced-stage cancer. Consequently, clinical testing of this cytokine-encoding mRNA mixture has recently been initiated by BioNTech in collaboration with Sanofi (NCT03871348).
4. In vitro-transcribed mRNA Chimeric Antigen Receptor T Cells
Chimeric antigen receptor T cells (CAR-T cells) are T cells that have been genetically engineered to produce an artificial T cell receptor for use in immunotherapy (Figure 2). Chimeric antigen receptors (CARs) are chimeric because they combine both antigen-binding and T-cell activating functions into a single receptor, thus giving these T cells the new ability to target a specific protein.
CAR-T cell therapy uses T cells engineered with CARs for cancer therapy. The premise of CAR-T immunotherapy is to modify T cells to recognize cancer cells to more effectively target and destroy them. Scientists harvest T cells from a cancer patient, genetically alter them, and then infuse the resulting CAR-T cells back into the patient to attack their tumor (Figure 2).
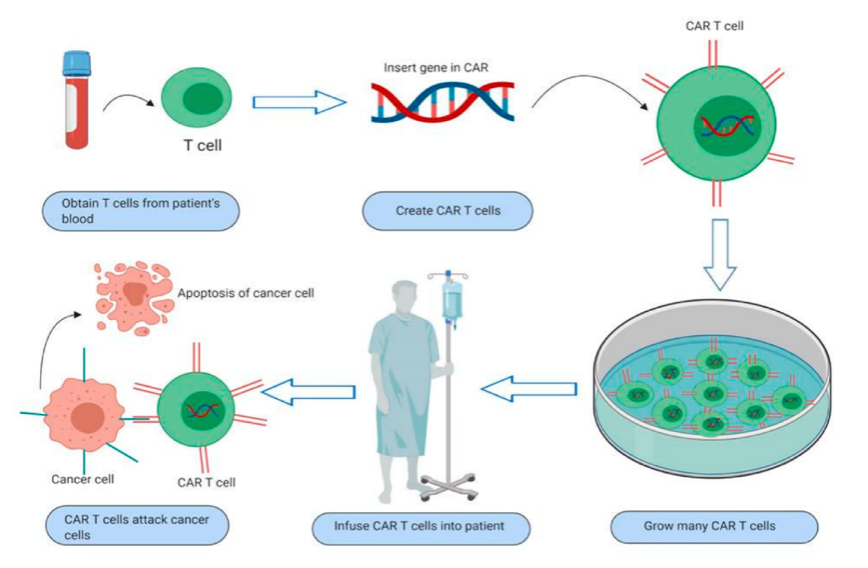
FIGURE 2. Schematic diagram of steps in CAR-T cell immunotherapy. Taken from Sur et al. and free to use under Creative Common CC BY license.
CAR-T cells have been clinically effective in killing certain hematological malignancies, but achieving long-term patient responses for solid tumors remains a challenge. A novel two-part strategy to overcome poor CAR-T cell stimulation and responses in vivo has been recently reported (Reinhard et al.).
First, Reinhard et al. studied in vivo antitumor activity of human CLDN6-CAR-T cells in mice xenografted subcutaneously with a human tumor cell line. Notably, all CLDN6-CAR-T cell-treated mice experienced complete tumor regression within 2 weeks, compared with control group mice with tumors that progressed rapidly. The second part is based on the group’s previous studies (Kranz et al. 2016), showing that intravenously administered liposomal antigen-encoding mRNA (termed RNA-LPX) stimulates tumor-associated T cells in the natural repertoire of cancer patients. RNA-LPX delivers antigen to antigen-presenting cells in the spleen, lymph nodes, and bone marrow, and concomitantly initiates an immune-stimulatory program, promoting priming and robust expansion of antigen-specific T cells. Reinhard et al. tested whether this approach could be adapted to act as a CAR-T cell-amplifying RNA vaccine in further mouse models of lung and ovarian cancer.
Based on the findings, BioNTech announced in November 2021 that CLDN6-CAR-T cells (termed BNT211) are being investigated both as a monotherapy and in combination with CLDN6-LPX CAR-T (CARVac). Their first-in-human Phase 1/2 clinical trial (NCT04503278) evaluates the safety and preliminary efficacy in patients with CLDN6-positive relapsed or refractory advanced solid tumors. In initial findings (Mackensen et al. 2021), eight patients had been treated and, “strikingly, three patients showed initial tumor shrinkage” (-18%, -21%, and -27%).
As for Moderna, it was reported in January 2022 that the company forged a partnership with Carisma Therapeutics to develop and commercialize in vivo engineered CAR monocyte (CAR-M) therapeutics for different forms of cancer. The research collaboration will combine Carisma’s engineered macrophage technology with Moderna’s mRNA and LNP technologies to generate and develop in vivo CAR-M therapeutics aimed at up to 12 targets.
Promising Pipelines
Many other preclinical and clinical immuno-oncology studies are being pursued by various companies with healthy mRNA therapeutics pipelines. As with all drug development, not all of these efforts will be successful, but some likely will be—and that is the important takeaway message.
It’s encouraging to note that President Biden announced on February 2, 2022, that his administration has ‘reignited’ his 2016 Cancer Moonshot program. Based on the successes of mRNA vaccines against COVID-19, the program will include funding the development of mRNA vaccines to fight cancer.
Your comments are welcomed, as usual.
Please feel free to share this blog with your colleagues or on social media.




