mRNA Basics
The human body can produce thousands of different proteins, serving varied functions including catalysis, structural, and signaling. Instructions for synthesizing these proteins are stored in DNA, the genetic code. However, proteins cannot be synthesized directly from DNA. An intermediate is required to carry instructions from DNA in the nucleus out to the cytoplasm, where proteins are produced.
Messenger RNA, also known as mRNA, is the link between DNA in the nucleus and protein synthesis machinery in the cytoplasm. mRNA is an ordered chain of nucleoside triphosphate bases, and also contains a polyadenylated tail (poly A tail) and 5’ cap structure. Through the process of transcription, a pre-mRNA strand is produced by RNA polymerase from a DNA template in the nucleus. Capping, polyadenylation, and splicing in the nucleus convert the pre-mRNA into fully processed mRNA. This mRNA transcript is a copy of the genetic code required to synthesize the specific protein of interest. Three-base units in the mRNA, called codons, specify which amino acids the final protein will be composed of.
Processed mRNA is exported from the nucleus and travels to the cytoplasm, where it engages with ribosomes for translation. During translation, amino acids are linked together in the order specified by the mRNA to form the desired protein. Over time, the poly A tail is shortened by exonucleases and eventually the mRNA is de-capped and degraded.
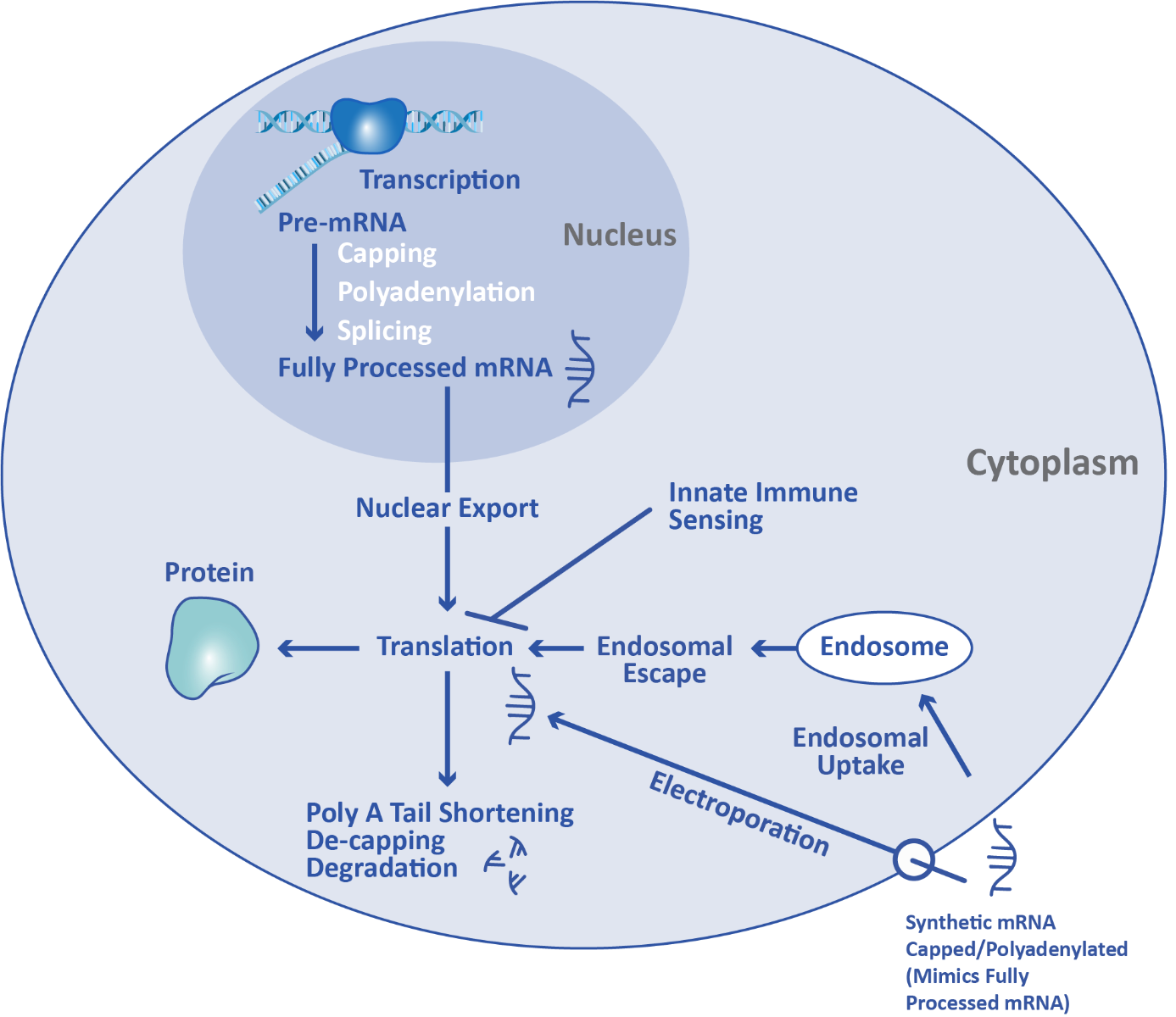
mRNA therapeutics take advantage of the cell’s existing translation machinery to produce proteins. Synthetic mRNA produced through in vitro transcription is introduced directly into the cytoplasm through a variety of methods, such as lipid nanoparticle transfection or electroporation. This externally manufactured mRNA uses the same ribosome machinery as mRNA produced in the cell, enabling it to be translated into the protein of interest.
However, in order for synthetic mRNA to successfully be translated in vivo, it must evade detection by the innate immune system. Unlike the adaptive immune system which detects specific invaders, the innate immune system operates by identifying common signals of damage or infection. These motifs are called Danger-Associated Molecular Patterns (DAMPs) and Pathogen-Associated Molecular Patterns (PAMPs), depending if they are found in the host environment or in pathogens. Pattern Recognition Receptors (PRRs) bind to DAMPs/PAMPs and trigger a downstream immune response.
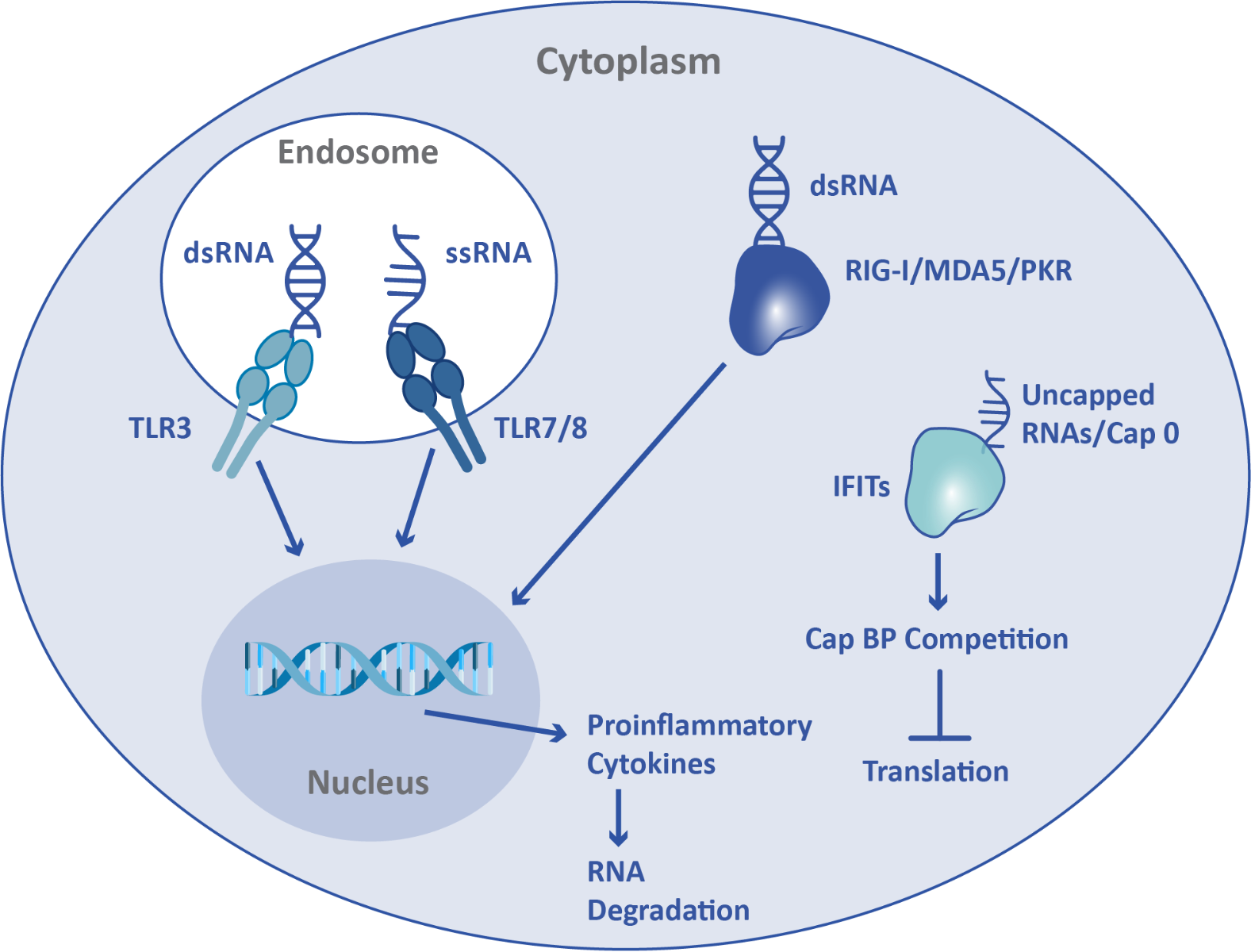
Since the innate immune system protects against viral infection, mRNA can function as a DAMP. Activation of the innate immune system by exogenous mRNA leads to inflammation, inhibition of translation, and mRNA degradation. When present in endosomes, single-stranded RNA, including mRNA, is recognized as a DAMP by Toll-Like Receptor (TLR) 7 and TLR8. Double-stranded RNA is recognized both in endosomes and in the cytoplasm, by TLR3 and retinoic acid-inducible gene I (RIG-I)/melanoma differentiation-associated protein 5 (MDA5)/protein kinase R (PKR) respectively. Additionally, interferon-induced tetratricopeptide repeat (IFIT) proteins can recognize aberrant mRNA cap structures. In order to avoid detection by the innate immune system, and thus enable translation, mRNA can be sequence-optimized, chemically modified, and capped.
Altering the mRNA’s base composition is an established method of reducing immunogenicity for mRNA therapeutics. Since uridine-rich sequences of RNA trigger the innate immune response, depleting uridine triphosphate from the final mRNA sequence can improve evasion. Sequence optimization takes advantage of synonymous codons, which are different codons that encode the same amino acid. Substituting a U-containing codon for one without will result in the same downstream protein while reducing the mRNA immunogenicity. Similarly, modified nucleoside triphosphates (NTPs) such as pseudouridine triphosphate, N1-methylpseudouridine triphosphate, and 5-methoxyuridine triphosphate can be substituted for the uridine triphosphate base to evade innate immune detection. Many mRNA therapeutics take advantage of both synonymous codons for uridine depletion and modified NTPs.
Mature mRNA produced in higher eukaryotes contains a 5’ cap structure called Cap 1 that protects against the innate immune response, enabling greater translation in vivo. Cap 1 features a 7-methylguanosine (m7G) connected by triphosphate to the first nucleotide of the mRNA, as well as a methylated 2’ ribose on the first nucleotide. Cap 0 does not have the methylation on the first nucleotide, but does have the m7G. IFITs have a weaker binding affinity for Cap 1 in comparison to Cap 0, and Cap 1 has also been shown to prevent MDA5 detection of mRNA. mRNA therapeutics require a cap structure in order to successfully persist and be expressed in the cell. Cap 0 RNAs have been shown to express poorly in mouse liver in vivo.
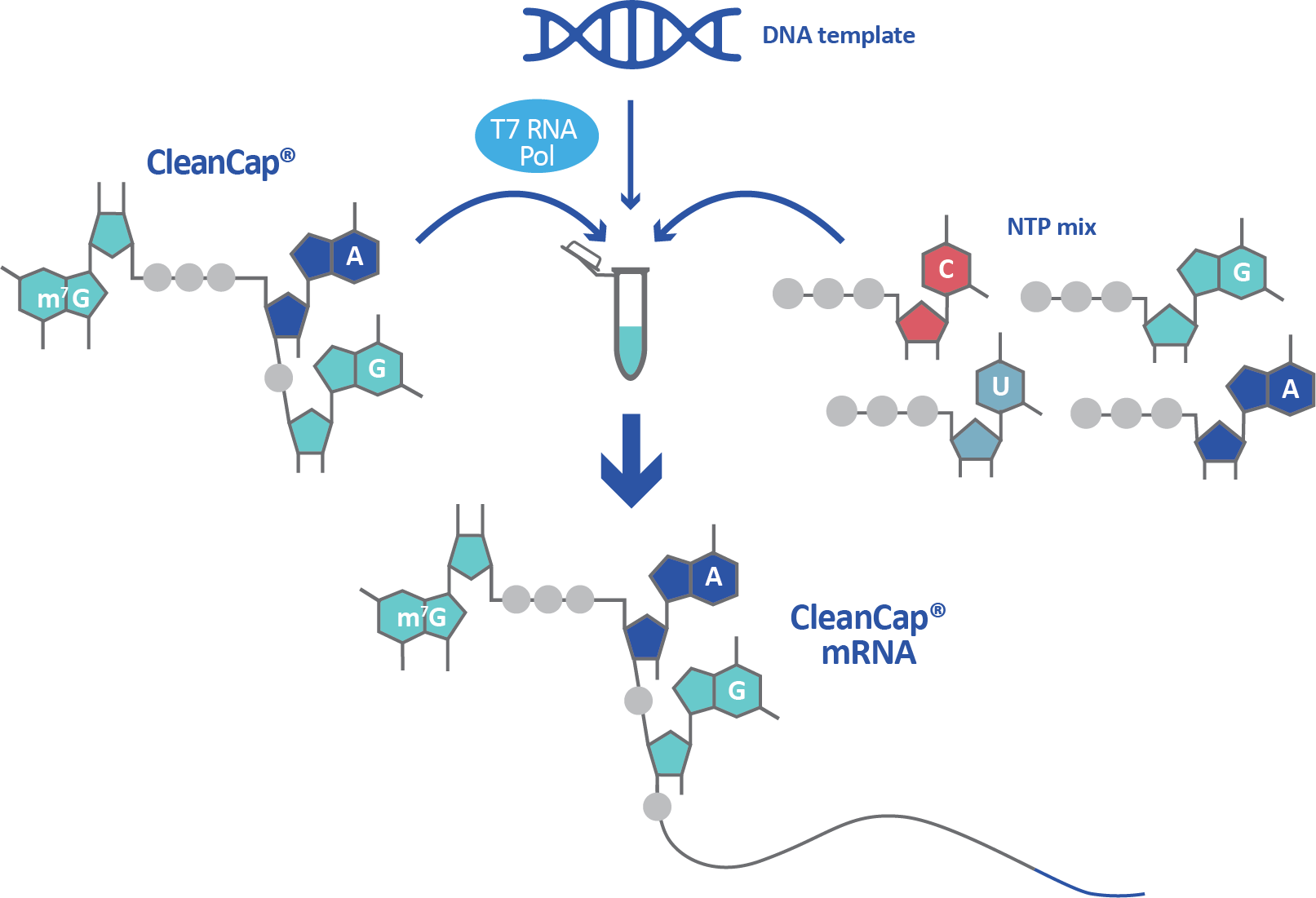
Synthetic mRNA is not capped by default and capping must be built into the manufacturing process. Co-transcriptional capping methods such as anti-reverse cap analog (ARCA) and CleanCap® have been developed to overcome this obstacle, as have post-transcriptional methods. Co-transcriptional capping is achieved by adding cap analogs like ARCA or CleanCap into the transcription reaction, where they are incorporated into the final mRNA. While CleanCap generates a Cap 1 structure, ARCA generates the less advantageous Cap 0 structure. Co-transcriptional capping with a Cap 1 structure is the most streamlined and effective manufacturing method for a high yield of non-immunogenic mRNA.